
3.2: Levels (Orders) of Protein Structure
- Page ID
- 16423

A. Primary Structure
1. L amino acids and the C-N-C-N-… polypeptide backbone
The primary structure of a protein refers to the amino acid sequence of its polypeptide chain(s). Cells use only 20 amino acids to make polypeptides and proteins, although they do use a few additional amino acids for other purposes. Peptide linkages between amino acids in polypeptides form in condensation reactions in cells during protein synthesis (i.e., translation). The linkages involve multiple covalent bonds. They break and rearrange between the carboxyl and amino groups of amino acids during linkage formation.
The 20 amino acids found in proteins are shown below.

Except for glycine, the a-carbon in the 19 other amino acids is bound to four different groups, making them chiral or optically active.
Recall that chiral carbons allow for mirror image D and L or d and l optical isomers. Recall also that only the lower case d or l defines the optical properties of isomers. Just to make life interesting, L amino acids are actually dextrorotary in a polarimeter, making them d-amino acids! While both enantiomers exist in cells, only dextrorotary d (i.e., L) amino acids (along with glycine) are used by cells to build polypeptides and proteins. A partial polypeptide is illustrated below.

The result of translation in a cell is a polypeptide chain with a carboxyl end and an amino end. Amino acid side chains (circled above) end up alternating on opposite sides of a C-N-C-N-… polypeptide backbone because of the covalent bond angles along the backbone. You could prove this to yourself by assembling a short polypeptide with a molecular modeling kit, the kind you might have used in a chemistry class! The C-N-C-N-…backbone is the underlying basis of higher orders, or levels of protein structure (see below).
2. Determining Protein Primary Structure - Polypeptide Sequencing
Frederick Sanger was the first to demonstrate a practical method for sequencing proteins when he reported the amino acid sequence of the two polypeptides of Bovine (cow) insulin. Briefly, the technique involves stepwise hydrolysis of polypeptide fragments, called an Edman Degradation. Each hydrolysis leaves behind a polypeptide fragment shortened by one amino acid that can be identified. Sanger received a Nobel Prize in 1958 for this feat. By convention, the display and counting of amino acids always starts at the amino-, or N-terminal end (the end with a free NH2-group). Primary structure is dictated directly by the gene encoding the protein. After transcription of a gene), a ribosome translates the resulting mRNA into a polypeptide.
For some time now, the sequencing of DNA has replaced most direct protein sequencing. The method of DNA sequencing, colloquially referred to as the Sanger dideoxy method, quickly became widespread and was eventually automated, enabling rapid gene (and even whole genome) sequencing. Now, instead of directly sequencing polypeptides, we can infer amino acid sequences from gene sequences isolated by cloning or revealed after complete genome sequencing projects. This is the same Sanger who first sequenced proteins, and yes…, he won a second Nobel Prize for the DNA sequencing work in 1980!
The different physical and chemical properties of amino acids themselves result from the side chains on their a-carbons. The unique physical and chemical properties of polypeptides and proteins are determined by their unique combination of amino acid side chains and their interactions within and between polypeptides. In this way, primary structure reflects the genetic underpinnings of polypeptide and protein function. The higher order structures that account for the functional motifs and domains of a mature protein derive from its primary structure. Christian Anfinsen won a half-share of the 1972 Nobel Prize in Chemistry for demonstrating that this was the case for the ribonuclease enzyme (Stanford Moore and William H. Stein earned their share of the prize for relating the structure of the active site of the enzyme to its catalytic function). See 1972 Nobel Prize in Chemistry for more.
B. Secondary Structure
Secondary structure refers to highly regular local structures within a polypeptide (e.g., a helix) and either within or between polypeptides (b-pleated sheets). Linus Pauling and coworkers suggested these two types of secondary structure in 1951. A little Linus Pauling history is would be relevant here! By 1932, Pauling had developed his Electronegativity Scale of the elements that could predict the strength of atomic bonds in molecules. He contributed much to our understanding of atomic orbitals and later to the structure of biological molecules. He earned the 1954 Nobel Prize in Chemistry for this work. He and his colleagues later discovered that sickle cell anemia was due to an abnormal hemoglobin, and went on to predict the alpha helical and pleated sheet secondary structure of proteins. While he did not earn a second Nobel for these novel molecular genetics studies, he did win the 1962 Nobel Peace prize for convincing almost 10,000 scientists to petition the United Nations to vote to ban atmospheric nuclear bomb tests. A more detailed review of his extraordinary life (e.g., at Linus Pauling-Short Biography) is worth a read!
Secondary structure conformations occur due to the spontaneous formation of hydrogen bonds between amino groups and oxygens along the polypeptide backbone, as shown in the two left panels in the drawing below. Note that amino acid side chains play no significant role in secondary structure.

133 Protein secondary structure
The a helix or b sheets are a most stable arrangement of H-bonds in the chain(s). These regions of ordered secondary structure in a polypeptide can be separated by varying lengths of less structured peptide called random coils. All three of these elements of secondary structure can occur in a single polypeptide or protein that has folded into its tertiary structure, as shown at the right in the illustration above. The pleated sheets are shown as ribbons with arrowheads representing N-to-C or C-to-N polarity of the sheets. As you can see, a pair of peptide regions forming a pleated sheet may do so either in the parallel or antiparallel directions (look at the arrowheads of the ribbons), which will depend on other influences dictating protein folding to form tertiary structure. Some polypeptides never go beyond their secondary structure, remaining fibrous and insoluble. Keratin is perhaps the best-known example of a fibrous protein, making up hair, fingernails, bird feathers, and even filaments of the cytoskeleton. Most polypeptides and proteins however, do fold and assume tertiary structure, becoming soluble globular proteins.
C. Tertiary Structure
Polypeptides acquire their tertiary structure when hydrophobic and non-polar side chains spontaneously come together to exclude water, aided by the formation of salt bridges and H-bonds between polar side chains that find themselves inside the globular polypeptide. In this way, a helices or b sheets are folded and incorporated into globular shapes. The forces that cooperate to form and stabilize 3-dimensional polypeptide and protein structures are illustrated below.

Polar (hydrophilic) side chains that can find no other side-chain partners are typically found on the outer surface of the ‘’globule’, where they interact with water and thus dissolve the protein (recall water of hydration). Based on non-covalent bonds, tertiary structures are nonetheless strong simply because of the large numbers of otherwise weak interactions that form them. Nonetheless, covalent disulfide bonds between cysteine amino acids in the polypeptide (shown above) can further stabilize tertiary structure. Disulfide bonds (bridges) form when cysteines far apart in the primary structure of the molecule end up near each other in a folded polypeptide. Then the –SH (sulfhydryl) groups in the cysteine side chains are oxidized, forming the disulfide (–S-S- ) bonds.
The sulfhydryl oxidation reaction is shown below.
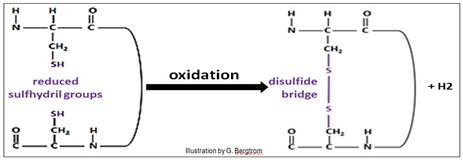
134 Protein Tertiary (30) Structure
135 Disulfide Bridges Stabilize 30 Structure
To better understand how disulfide bridges can support the 3-dimensional structure of a protein, just imagine its physical and chemical environment. Changing the temperature or salt concentration surrounding a protein might disrupt non-covalent bonds involved in the 3D shape of the active protein. Unaffected by these changes, disulfide bridges limit the disruption and enable the protein to re-fold correctly and quickly when conditions return to normal (think homeostasis!).