
9.3: DNA Repair
- Page ID
- 16466

A. DNA Damage (Mutation) is a Fact of Life
We generally accept the notion that replication faithfully duplicates the genetic material. At the same time, evolution would not be possible without mutation, and mutation is not possible without at least some adverse consequences.
Germline mutations are heritable. When present in one, but especially in both alleles of a gene, such mutations can result in genetic disease (e.g., Tay-Sach’s disease, cystic fibrosis, hemophilia, sickle-cell anemia, etc.). Rather than causing disease, some germline mutations may increase the likelihood of becoming ill (e.g., mutations of the BRCA2 gene greatly increase a woman’s odds of getting breast cancer). Somatic mutations in actively dividing cells might result in benign “cysts” or malignant tumors (i.e., cancer). Other somatic mutations may play a role in dementia (Alzheimer’s disease) or in some of the neuropathologies along the autism spectrum.
Since the complex chemistry of replication is subject to an inherent high rate of error, cells have evolved systems of DNA repair to survive high mutation rates. As we saw, DNA polymerases themselves have proofreading ability so that incorrectly inserted bases can be quickly removed and replaced. Beyond this, multiple mechanisms have evolved to repair mismatched base pairs and other kinds of damaged DNA that escape early detection. How often and where DNA damage occurs is random, as is which damage will be repaired and which will escape to become a mutation. For those suffering the awful consequences of unrepaired mutation, the balance between retained and repaired DNA, damage is to say the least, imperfect. However, evolution and the continuance of life itself rely on this balance.
B. What Causes DNA Damage
DNA is most exposed and therefore most vulnerable to damage, especially in eukaryotes. The simplest damage to DNA during replication is the point mutation, the accidental insertion of a ‘wrong’ nucleotide into a growing DNA strand. Other mutations, equally accidental, include DNA deletions, duplications, inversions, etc., any of which might escape repair. The causes of DNA damage can be chemical or physical, and include spontaneous intracellular events (e.g., oxidative reactions) and environmental factors (radiation, exogenous chemicals, etc.). Based on studies of different kinds of DNA damage, Thomas Lindahl estimated that DNA damaging events might be occurring at the rate of 10,000 per day! Lindahl realized that there must be some “fundamental DNA repair mechanisms” at work to protect cells against such a high rate of DNA damage. The discovery of the base excision repair mechanism earned Thomas Lindahl a share in the 2015 Nobel Prize in Chemistry. Environmental factors that can damage DNA include UV light, X-rays and other radiation, as well as chemicals (e.g., toxins, carcinogens, and even drugs, etc.). Both germline and somatic cells can be affected. While mutations can and do cause often debilitating diseases, it is instructive to keep the impact of mutations in perspective. Most mutations are actually silent; they do not cause disease. In addition, much DNA damage is repaired. Cells correct more than 99.9% of mistaken base changes before they have a chance to become mutations. That is why we think of replication as a “faithful” process. Let’s look at some common types of DNA damage that are usually repaired:
- Pyrimidine dimers, typical of adjacent thymines (less often cytosines) in a single DNA strand, caused by UV exposure
- Depurination; the hydrolytic removal of guanine or adenine from the #1 C (carbon) of deoxyribose in a DNA strand
- Deamination: hydrolytic removal of amino (-NH2) groups from guanine (most common), cytosine or adenine
- Oxidative damage of deoxyribose with any base, but most commonly purines
- Inappropriate methylation of any bases, but most commonly purines
- DNA strand breakage during replication or from radiation or chemical exposure
C. Some Molecular Consequences of Uncorrected DNA Damage
While bacteria suffer DNA damage, we will focus here on eukaryotes since they have evolved the most sophisticated mechanisms. Remember that unrepaired DNA damage will be passed on to daughter cells in mitosis, or might be passed on to the next generation if the mutation occurs in a germline cell.
Next, let us consider some molecular consequences of uncorrected DNA damage.
1. Depurination
This is the spontaneous hydrolytic removal of guanine or adenine from deoxyribose C#1 in a DNA strand. Its frequency of 5000 depurinations per cell per day emphases the high rate of DNA damage that demands a fix! If not repaired, depurination results in a single base-pair deletion in one chromosome after replication, leaving the DNA in the same region of the other chromosome unchanged. The effects of depurination are illustrated below.
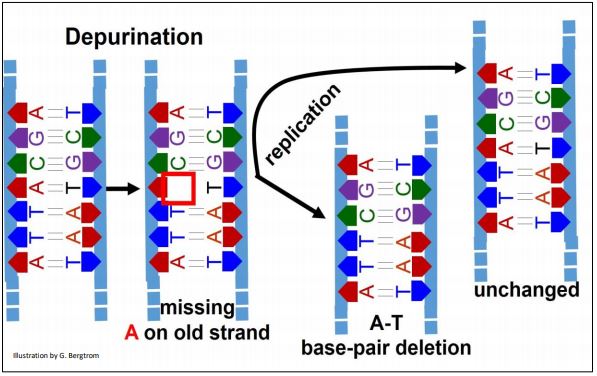
The replisome ignores the missing base during replication of the depurinated DNA region (an A in this example), jumping to the C in the depurinated template DNA. Unrepaired, one new double-stranded DNA will have a deletion, leaving the other new one with no mutation.
2. Pyrimidine Dimerization
UV light exposure of DNA can cause adjacent pyrimidines (commonly thymines; less often, cytosines) on a DNA strand to dimerize. Pyrimidine dimers form at a rate of a bit less than 100 per cell per day!
Uncorrected dimerization results in 2-base deletion in one chromosome while the other is unchanged (below).
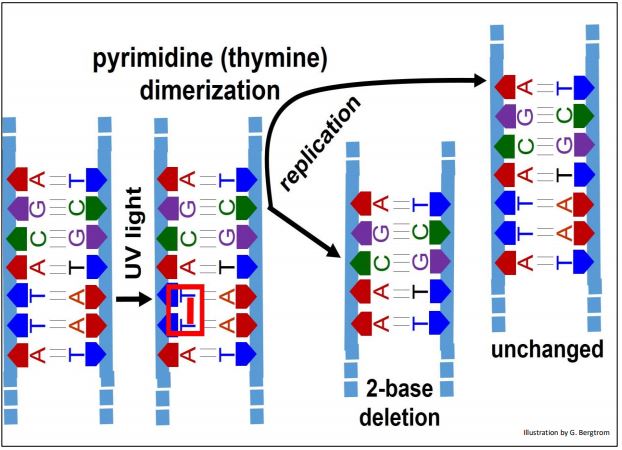
You can predict that correction of this radiation-induced damage will either involve disrupting the dimers (in this case thymine dimers), or removal and replacement of the dimerized bases by monomeric bases.
3. Deamination
Deamination is the hydrolytic removal of amino (-NH2) groups from guanine (most common), cytosine or adenine, at a rate of 100 per cell per day. Deamination does not affect thymine (because it has no –amino groups!). Uncorrected deamination results in a base substitution on one chromosome (actually, a T-A pair substitution for the original C-G in this example) and no change on the other. Deamination of adenine or guanine results in unnatural bases (hypoxanthine and xanthine, respectively). These are easily recognized and corrected by DNA repair systems. The U-A base pair remains occasionally un-repaired. The consequences of deamination to base sequence are shown below.

D. DNA Repair Mechanisms
Many enzymes and proteins are involved in DNA repair. Some of these function in normal replication, mitosis and meiosis, but were co-opted for DNA repair activities. These molecular co-optations are so vital to normal cell function that some repair activities and molecular players are highly conserved in evolution. Among different DNA repair pathways that have been identified, we will look at Base Excision Repair, Nucleotide Excision Repair, Transcription Coupled Repair, Non-homologous End-Joining, and Homologous Recombination (of these, the last is perhaps the most complex).
1. Base Excision Repair
Upon detection and recognition of an incorrect base (e.g., oxidized bases, openring bases, deaminated Cs or As, bases containing C=C bonds saturated to C-C bonds…), specific DNA glycosylases catalyze hydrolysis of the damaged base from affected deoxyribose in the DNA. To learn more about the specific versions of this enzyme, click here. Events of base excision repair are summarized below.

After a DNA glycosylase removes an offending base, an AP endonuclease recognizes the deoxyribose with the missing base and nicks the DNA at that nucleotide. Phosphodiesterase next hydrolyzes the remaining phosphate-ester bond of ‘base-less’ sugar phosphate, removing it from the DNA strand. DNA polymerase then adds correct nucleotide to the 3’ end of the nick. Finally, DNA ligase III (an ATP-dependent mammalian version of the original prokaryotic enzyme) seals the remaining nick in the strand. Thomas Lindahl (see above) discovered most of these enzymes.
2. Nucleotide Excision Repair
The discovery of nucleotide excision repair earned Aziz Sancar a share in the 2015 Nobel Prize in Chemistry. The results of this mechanism include the removal of thymidine dimers. The events of nucleotide excision repair are shown below for a pyrimidine dimer.
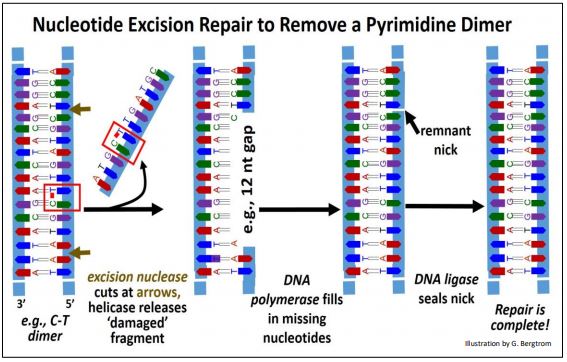
In this example, an Excision Nuclease recognizes a pyrimidine dimer and hydrolyzes phosphodiester bonds between nucleotides several bases away from either side of the dimer. A DNA helicase then unwinds and separates the DNA fragment containing the dimerized bases from the damaged DNA strand. Finally, DNA polymerase acts 5’-3’ to fill in the gap and DNA ligase seals the remaining nick to complete the repair.
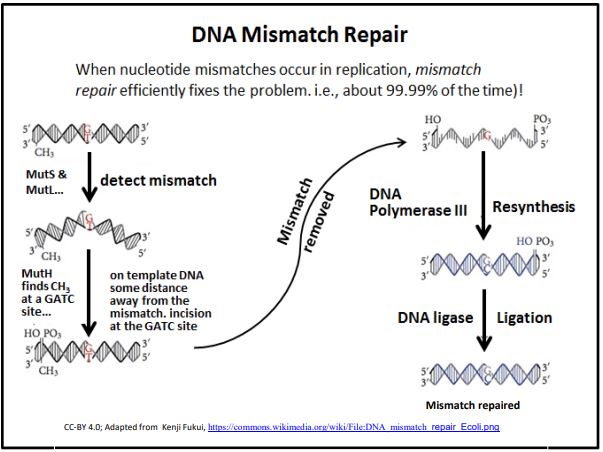
3. Mismatch Repair
DNA Mismatch Repair occurs when DNA polymerase proofreading misses an incorrect base insertion into a new DNA strand. This repair mechanism relies on the fact that double-stranded DNA shows a specific pattern of methylation. The discovery of the mismatch repair mechanism earned Paul Modrich a share in the 2015 Nobel Prize in Chemistry. These methylation patterns are related to epigenetic patterns of gene activity and chromosome structure that are expected to be inherited by daughter cells. When DNA replicates, the methyl groups on the template DNA strands remain, but the newly synthesized DNA is unmethylated. In fact, it will take some time for methylation enzymes to locate and methylate the appropriate nucleotides in the new DNA. In the intervening time, several proteins and enzymes can detect inappropriate base pairing (the mismatches) and initiate mismatch repair. The basic process is illustrated below.
4. Transcription Coupled Repair (in Eukaryotes)
If an RNA polymerase reading a template DNA encounters a nicked template or one with an unusual base substitution, it might stall transcription and “not know what to do next”. Thus at a loss, a normal transcript would not be made and the cell might not survive. No big deal in a tissue comprised of thousands if not millions of cells, right? Nevertheless, Transcription Coupled Repair exists! In this repair pathway, if RNA polymerase encounters a DNA lesion (i.e., damaged DNA) while transcribing a template strand, it will indeed stall. This allows time for coupling proteins to reach the stalled polymerase and enable repair machinery (e.g., by base, or nucleotide excision) to effect the repair. Once the repair is complete, the RNA polymerase ‘backs up’ along the template strand with the help of other factors, and resumes transcription of the corrected template.
5. Non-homologous End-Joining
DNA replication errors can cause double stranded breaks, as can environmental factors (ionizing radiation, oxidation, etc.). Repair by non-homologous end-joining deletes damaged and adjacent DNA and rejoins the ‘cut’ ends (shown below).
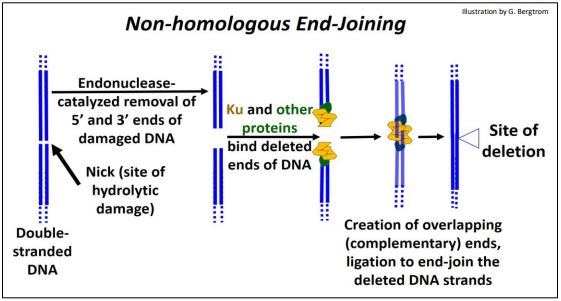
Once the site of a double-stranded break is recognized, nucleotides hydrolyzed from the ends of both strands at the break-site leave ‘blunt ends’. Next, several proteins (Ku among others) bring DNA strands together and further hydrolyze single DNA strands to create staggered (overlapping, or complementary) ends. The overlapping ends of these DNA strands form H-bonds. Finally, DNA ligase seals the H-bonded overlapping ends of DNA strands, leaving a repair with deleted bases.
In older people, there is evidence of more than 2000 ‘footprints’ of this kind of repair per cell. How is this possible? This quick-fix repair often works with no ill effects because most of the eukaryotic genome does not encode genes or even regulatory DNA (whose damage would otherwise be more serious).
6. Homologous Recombination
Homologous recombination is a complex but normal and frequent part of meiosis in eukaryotes. You may recall that homologous recombination occurs in synapsis in the first cell division of meiosis (Meiosis I). During synapsis,homologous chromosomes align. This may lead to DNA breakage, an exchange of alleles, and ligation to reseal the now recombinant DNA molecules. Novel recombinations of variant alleles in the chromosomes of sperm and eggs ensure genetic diversity in species. The key point is that DNA breakage of DNA is required to exchange alleles between homologous chromosomes. Consult the genetics chapter in an introductory biology textbook, or the recombination chapter in a genetics text to be reminded of these events.
Cells use the same machinery to reseal DNA breaks during normal recombination and to repair DNA damaged by single or double stranded breakage. A single DNA strand nicked during replication can be repaired by recombination with strands of homologous DNA that are being replicated on the other strand. A double stranded break can be repaired using the same recombination machinery that operates on sister chromatids in meiosis. In both cases, the process accurately repairs damaged DNA without any deletions. These mechanisms are conserved in the cells of all species. This indicates an evolutionary imperative of accurate repair to the survival of species, no less than the imperative to maintain genetic diversity of species.
a) Repair of a Single-Stranded Break
A specific example of homologous recombination repair is the re-establishment of a replication fork damaged when a replisome reaches a break in one of the two strands of replicating DNA (illustrated below).
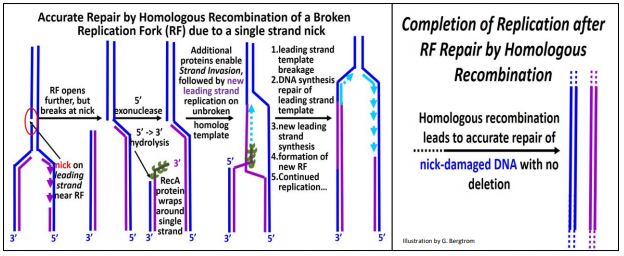
Such a break may have occurred prior to replication itself, and repair begins when the replication fork (RF) reaches the lesion. In the first step, a 5’-3’ exonuclease trims template DNA back along its newly synthesized complement. Next, RecA protein monomers (each with multiple DNA binding sites) bind to the single-stranded DNA to form a nucleoprotein filament. With the help of additional proteins, the 3’ end of the ‘filament’ scans the ‘other’ replicating strand for homologous sequences. When such sequences are found, the RecA-DNA filament binds to the homologous sequences and the filament of new DNA ‘invades’ the homologous (i.e., opposite) double stranded DNA, separating its template and newly replicated DNA. After strand invasion, replication of a leading strand continues from the 3’ end of the invading strand. A new RF is established as the leading strand template is broken and re-ligated to the original break; New lagging strand replication then resumes at the new (re-built) RF. The result is an accurate repair of the original damage, with no deletions or insertions of DNA.
RecA, a bacterial protein, is another of those evolutionarily conserved proteins. Its homolog in Archaea is called RadA. In Eukaryotes, the homolog is called Rad51, where it initiates synapsis during meiosis. Thus, it seems that a role for RecA and its conserved homologs in DNA repair predated its use in synapsis and crossing over in eukaryotes! For more about the functions of RecA protein and its homologs, click here.
b) Repair of a Double-Stranded Break
Homologous recombination can also repair a double-stranded DNA break with the aid of a number of enzymes and other proteins. Alternate repair pathways are summarized in the illustration on the next page. Here is a list of proteins involved in these homologous recombination pathways:
MRX, MRN: bind at double-stranded break; recruit other factors.
Sae2: an endonuclease (active when phosphorylated).
Sgs1: a helicase.
Exo1, Dna2: single strand exonucleases.
RPA, Rad51, DMC1: proteins that bind to overhanging DNA to form a nucleoprotein filament and initiate strand invasion at similar sequences.
The activities of other enzymes in the drawing are identified. Not shown in this illustration are two gene products that interact with some of the proteins that mediate the repair pathway. These are products of the BRCA1 and BRCA2 genes (the same ones that when mutated, increase the likelihood of a woman getting breast cancer). Expressed mainly in breast tissue, their wild-type (normal) gene products participate in homologous recombination repair of double-stranded DNA breaks. They do this by binding to Rad51 (the human RecA homolog!).

When mutated, the BRCA proteins function poorly and DNA in the affected cells is not efficiently repaired. This is the likely basis of the increased chance of getting breast cancer. It doesn’t help matters that the normal BRCA1 protein also plays a role in mismatch repair… and that the mutated protein can’t! To end this chapter, here is a bit of weird science! Read all about the genome of a critter, nearly 17% of which is comprised of foreign DNA, possibly the result of here.