
6.3: Different Drug Application Review Processes
- Page ID
- 39508

New Drug Application (NDA)
If the drug passes all three phases of testing, the company may file a New Drug Application (NDA), which permits the FDA to ascertain that the new drug (or biologic) is safe, reliable, and effective for the indications on the labeling. The FDA employs expert reviewers who examine the test results and determine whether the new drug can be approved.
Types of Drug Applications:
- Traditional 505(b)(1) NDA – FD&C Act, Section 505(b)(1)
- 505(b)(2) NDA
- Abbreviated NDA (ANDA) 505(j)
- Original BLA – PHS Act, Section 351(a) – for biologics discussed in the next chapter
- Biosimilar BLA – PHS Act, Section 351(k) – for biosimilars discussed in the next chapter
New Application of the Same Drug - Review Process
Approval of a new application of the same drug: If a company wants to expand the use of their drug to a new application, previous safety studies are usually still applicable; however, new clinical studies will be required to test the efficacy of the drug for its new application. The 505(b) (1) NDA is the complete application with all the appropriate study information outlined in the CFR that will demonstrate the drug’s safety and effectiveness. The 505(b) (2) NDA can include drugs where safety and effectiveness have been established in previous studies (by other companies), allowing companies to develop treatments quicker with less clinical study volunteers. An example of an application would be instead of a ten-day treatment of the drug, the drug now has a slow-release capsule, so it has only 3-day treatment, but is slowly released over ten days.
Abbreviated New Drug Application (ANDA)
Interestingly, the term ‘generic drug' is not defined in FDA regulations. A generic drug must have the same active ingredient, same potency, and the same dosage to be sold without having to repeat the extensive clinical trials used in the development of the original, brand-name drugs. Generics must deliver the same amount of active ingredient into a patient's bloodstream over the same period as the brand name – this is referred to as bioequivalence. The rate and extent of absorption of a drug are called its bioavailability. If the bioavailability of the two is similar, the drugs are bioequivalent.
An ANDA (505(j)), is an application for approval for a bioequivalent drug product. This application contains data submitted to FDA's Center for Drug Evaluation and Research, Office of Generic Drugs for their review for approval. Generic drug applications are called "abbreviated" because they frequently are not required to include any preclinical (animal) or clinical (human) data to show efficacy and safety. Instead, a generic applicant must scientifically demonstrate that its product has therapeutic equivalence. Therapeutic Equivalence means the drug must have the same clinical effect and safety (under the same labeling) as the non-generic drug and must have the identical active ingredient, with identical strength, quality, purity, and potency. It does not need to have the same inactive ingredients. Once approved, the company can then manufacture and market the generic drug product to provide a low-cost alternative to the brand-name drug.
Biological License Application (BLA)
A BLA is required for biological products submitted to CBER or CDER (characterized protein). The BLA must include all safety and efficacy information necessary for drug approval. A 351(a) application (Original BLA), contains all the information required and outlined in 21 CFR 601.2. A 351(k) application is an abbreviated BLA for a biosimilar. Although some biologics are overseen by CDER, the BLA process is further explored in the Biologics Regulatory Approval chapter.
Expedited Reviews
The FDA identifies four expedited approval processes: fast track, breakthrough therapy, priority review, and accelerated approval. These expedited processes can help drugs get to the hands of consumers faster based on their tangible needs, such as treating a serious illness with a new drug or one that is substantially improved over current therapies. Learn more here: www.fda.gov/patients/learnabout-drug-and-device-approvals/fast-track-breakthrough-therapy-accelerated-approval-priority-review
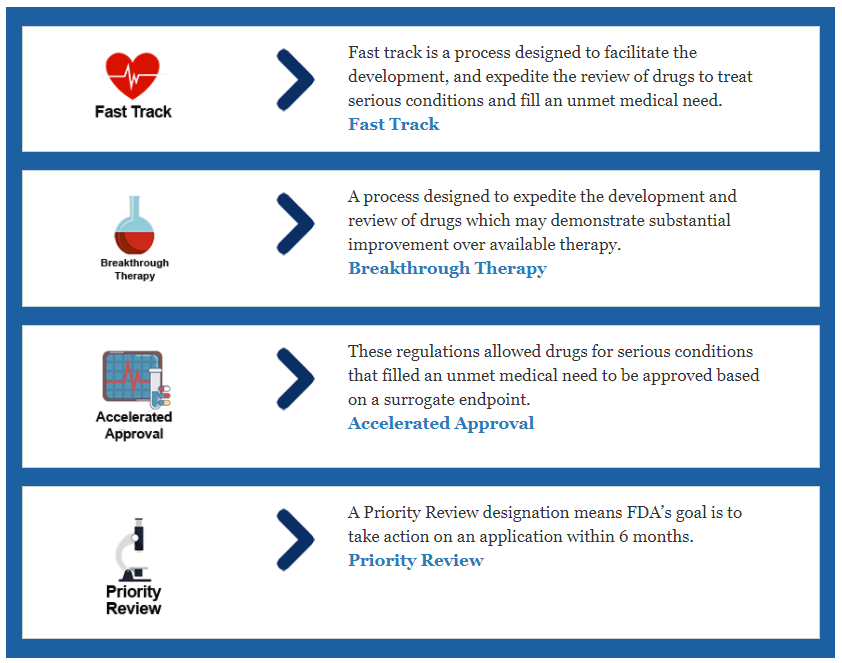
- Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need to ensure vital new drugs to the patient earlier. Fast Track addresses a broad range of serious conditions.
- Breakthrough Therapy Designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s).
- Priority Reviews These products represent significant improvements in the safety or efficacy of the treatment of a serious condition compared with currently marketed products.
- Accelerated Approval This program ensures products for serious conditions are made available earlier in the development process by relying on a surrogate endpoint that may predict a clinical benefit, such as a tumor shrinkage may mean increased survival.
Special Drug Incentive Program
There are several sponsored programs to encourage the development of a drug that is in demand. The Orphan Drug Act and GAIN (Generating Antibiotic Incentives Now) and the Presidential Emergency Plan for AIDS Relief (PEPFAR) are three examples of special drug incentive programs available. GAIN is an incentive program to develop drugs that treat life-threatening bacterial infections. GAIN drugs get Fast Track and Priority review in addition to 5-year market exclusivity (7 yrs. for orphan drugs!). To learn more about the FDA Safety and Innovation Act visit: www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/F DASIA/ucm20027187.htm
The FDA does permit the approval of drugs and vaccines intended to counter biological, chemical, and nuclear terrorism without first proving their safety and worth in Phase II and III trials. It would be unethical to deliberately expose humans to harmful radiation or pathogens to test the effectiveness of treatment. For example, the FDA expedited approval of one new drug, Cipro®, an antibiotic that adequately treats those who are exposed to anthrax. The FDA also has a more streamlined process for approval of "orphan drugs" (drugs with small numbers of beneficiaries but with great benefit).
Orphan Drugs
The FDA administers a program that provides incentives to develop drugs for use in patient populations of rare diseases (200,000 or fewer cases), or if there is a reasonable expectation that the drug will not be developed without FDA assistance. To learn more about rare diseases, see the Office of Rare Diseases Research: http://rarediseases.info.nih.gov/. Companies manufacturing orphan drugs receive the following inducements: seven-year marketing exclusivity, a tax credit for the productassociated clinical research, research design assistance from FDA, and grants
Test Your Knowledge!
Download and read Case Study 1: Drug Approval: Bringing a New Drug to the Market. We will review this case study https://www.fda.gov/media/94428/download
- In the case study, Dr. Green has hired regulatory affairs expert Dr. Robert’s, to guide the company’s development and FDA approval of Lowagliflozin, an NME to treat type 2 diabetes. What is an NME, and why does Lowagliflozin qualify as an NME?
- Explain where Lowagliflozin is in the approval process and the remaining milestones to go.
- Dr. Green asks the consultant if it is possible to do an expedited review for Lowagliflozin. What is an expedited review? What are the four expedited review programs that the FDA has? What do you think the chances are of obtaining expedited review?
- Once Lowagliflozin gets FDA approval, is that the end of their interaction with the FDA? Explain.
Over-The-Counter (OTC) Drug Review Process
Over-the-counter (OTC) drug products are drugs that can be obtained and used without a prescription. More than 300,000 OTC drug products are available in the US. OTC drugs are still overseen by CDER to ensure they are appropriately labeled, and benefits to their use outweigh the risks. To be designated OTC, the drugs must be considered safe, benefits outweigh the risks, and have minimal potential for abuse.
OTC drugs can still carry risks, in particular, risks of side effects, drug interactions, or overdose. One example is Tylenol (active ingredient acetaminophen), which most people assume is very safe. However, an overdose of this OTC drug results in over 44,000 individuals in the emergency room and over 400 die each year of liver failure. Learn more here! https://www.sciencedaily.com/releases/2015/06/150622124713.htm
Behind the Counter
Some OTC drugs are technically kept ‘behind the counter’ such as emergency contraceptive, and pseudoephedrine. Customers are intentionally prevented from direct access to these drugs due to their risk of unintentional or intentional misuse, as with some cold medicines that contain pseudoephedrine. Although a prescription is not required for pseudoephedrine, the pharmacist can dispense only with age and application verification.
OTC Drug Development and Review
Although many drugs are approved for OTC use through the new drug application (OTC NDA) review process, other OTC medicines are regulated under the OTC Monograph. This process relies on published monographs, which outline acceptable ingredients, doses, formulations, and consumer labeling for OTC drugs. Products that conform to a final monograph, and are generally recognized safe and effective (GRASE), may be marketed without prior FDA clearance.
Drugs that are not GRASE may be subject to FDA pre-approval through an NDA either through a direct-to-OTC NDA or prescription-to-OTC designation change. A direct-to-OTC NDA will require the same drug approval process as a prescription drug, except it may need a wider clinical testing population. The cold-sore OTC drug Abreva (docosanol) is an example of a direct-to-OTC application. A more common route is the prescription-to-OTC switch after a patent expires. This route is popular because of the Hatch-Waxman Act, which qualifies companies for a 3-yr extension of patent exclusivity. A prescription-to-OTC route requires a demonstration that the need of doctor oversight is unnecessary; the patient can self-diagnose, and the product has low toxicity.
In summary, all OTC drugs must comply with the same regulatory requirements and cGMP regulations for pharmaceuticals, as outlined in 21 CFR 210 and 211. Manufacturing sites must also register with the FDA, be subject to routine inspections, and be drug listed. However, OTC drugs undergo additional scrutiny to ensure they can be used safely and effectively by consumers without medical provider supervision.