
6.2: The Drug Development Process
- Page ID
- 39506

Overview
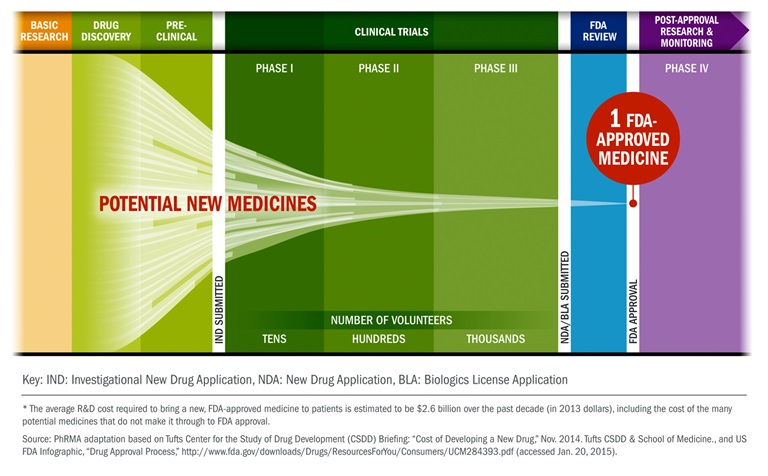
New medicines can take upwards of 12 years and costs $2.6 billion (Phrma, 2015). The first milestone for any new drug occurs during the research and discovery phase. Some form of experimentation in an R&D lab leads to the development of an active pharmaceutical ingredient (API) that may have therapeutic activity in the human body. If the active ingredient is believed to have real potential, the drug moves into the development stage. This early stage of development involves what is known as "preclinical testing," and is carried out in laboratories and animal testing facilities. This preclinical phase must be successful before testing can happen in humans. If the drug performs well during this stage, the company can file an Investigational New Drug Application (IND) with the FDA to request permission to begin testing on human subjects. There are three major phases a drug must pass through during human subject testing (clinical studies). If the drug passes through clinical studies successfully, the company can then submit a New Drug Application (NDA) to the FDA. If the FDA grants its approval, the company can finally begin selling the product, but its involvement does not end there. The FDA will continue to interact with the company to help ensure that the product is manufactured safely. It is important to note that the company that sponsors drug development may not be the company performing the tasks of the development. Contract Research Organizations (CROs) are frequently enlisted and paid to perform specific tasks of drug development. These tasks may include animal testing, human testing, and actual manufacturing.
Explore!
The FDA provides a simple overview of the drug review process here.
Do over-the-counter medications go through the same approval process?
Milestones in the Manufacture of a Drug
Research and Development
The earliest approaches to drug discovery involved identifying and isolating the active components of naturally occurring chemicals, such as those found in plants and other homeopathic remedies. In the modern laboratory, researchers also search for the causative agents of diseases (e.g., missing or over-active proteins) to guide drug research by understanding what problems must be targeted. High Throughput Screening (HTS) is another approach, which enables scientists to screen thousands of potential drugs all at once and allows for quick identification and targeting of potentially useful compounds.
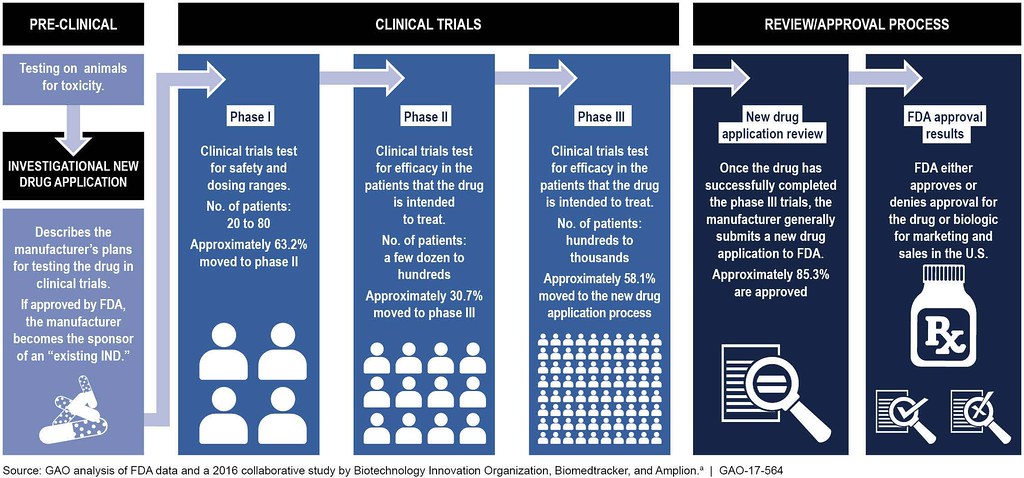
Preclinical Development
Before testing a drug in humans, it must undergo nonclinical testing to obtain basic toxicity and pharmacological data. Nonclinical testing must include animal models and assays to explore pharmacology, toxicity, reproductive toxicity, and genotoxicity. Preclinical development can take anywhere from 1-4 years and may require further testing to be in conjunction (or in parallel) with clinical studies.
The Objectives of Preclinical Development:
- Identifying the physical and chemical properties of the candidate drug
- Testing the candidate drug in vitro
- Determining formulation for administration to test subjects and patients
- Developing manufacturing methods for the candidate drug
- Testing the candidate drug in cultured cells
- Testing the candidate drug in animals for safety
- Developing analytical assays
- Securing intellectual property protection for the potential product, its uses, and its manufacture
ADME
For this potential drug to be useful, it must be stable, safe, and be manufactured practically. This stage is also dedicated to determining the drug's activity, chemical attributes, and solubility and outlining manufacturing schemes to ensure its potential as a drug. If the drug shows potential in the laboratory, the next step requires toxicity tests. These tests are also known as ADME (Absorption, Dissemination, Metabolic, and Excretion) studies. ADME studies are carried out on animals and help researchers determine:
- How much of the drug is absorbed by the blood?
- How is the substance metabolically altered in the body?
- What are the toxicity effects of metabolic by-products?
- How quickly will the drug and its by-products be excreted?
Toxicity
Safety assessment is done using toxicity studies. These studies are conducting using GLP guidelines for 30-90 days, in a minimum of two mammalian species, one of which must be non-rodent. The dosage, length of study, and complexity of study are related to the proposed clinical study; duration and complexity should be equal to or exceed what is proposed in humans. Additionally, if the new drug is also a New Chemical Entity (NCE) and has no long-term human data at all, the study may be required to exceed 12 months.
- Reproductive Toxicity. Fertility and embryonic development are also studied extensively in human clinical trials. This includes early embryonic development, embryo-fetal development, as well as pre and post-natal development.
- Genotoxicity. Genotoxicity, the propensity to damage genetic information, is also extensively studied in both in-vitro and in vivo. This assessment of mutagenicity is tested in both bacteria and mammalian cells.
- Carcinogenicity. Carcinogenicity studies are not required before clinical studies begin and may not have to be done for some products. These studies may take upwards of 2 years to complete.
Investigational New Drug Application (IND)
If the drug candidate is promising in the preclinical testing, then the company compiles its data and submits a plan to test the drug on human subjects to the FDA, called the Investigational New Drug Application (IND). The IND contains information from animal studies, information relating to the composition and manufacture of the drug, and the investigational plan. The IND application includes a description of the product, the results of animal tests, and the plans for further testing. The FDA then decides whether the company’s materials are sufficiently complete that the company can begin testing the product in humans.
The IND is not ‘approved'; rather, it becomes active within 30 days of the FDA receiving it. If deficiencies are discovered, the company is given an opportunity to correct it. If the issues are not addressed, the FDA will put the clinical studies on hold until they are. Some areas of concern for the FDA include unreasonable risk to human health, investigators without the appropriate credentials, and incomplete (or misleading) preclinical data.
IND Amendments
During the clinical development, the IND must be updated if any changes are made. These amendments may include changes to protocols, new toxicology data from animal studies that extended into the clinical studies, any adverse events, and any new findings that reveal this drug may cause a significant health risk for human volunteers.
Clinical Development
The government has an interest in protecting the public from defective products and drugs. Therefore, companies must demonstrate their effectiveness and safety before mass distribution. However, the only way they can actively do this is by having human subjects test out their products. As a refresher from the previous chapter on clinical studies, watch this video on clinical trials: youtu.be/pm1igf85uoA
Clinical Trials Data Bank
As previously discussed, all clinical data is published in the clinical trials data bank at clinicaltrials.gov, which is maintained by the National Institutes of Health (NIH). Companies are required to submit their Phase 2 and 3 clinical data. A company may request to withhold this data if they can prove that it will interfere substantially with a timely clinical study; however, this is up to the FDA.