
27.2: Regulation of Gene Expression in Eukaryotes
- Page ID
- 15207


As seen in Chapter 26, the initiation of transcription requires the assembly of a multitude of transcription factors (TF) localized at the promoter region. Transcription can also utilize far-reaching interactions of enhancers, that bind at a distant DNA site and loop back around to stabilize the RNA polymerase at the promoter. Control of transcriptional initiation is dependent on TF factor activation, TF binding with specific DNA recognition sequences, and chromatin remodeling.
Transcription Factor (TF) Activation
Many TFs are expressed within cells and held in an inactive conformation until the right environmental stimulus is present within the cell. Cellular signaling pathways can cause post-translational protein modifications leading to TF activation or small molecules may physically bind and allosterically modify the protein structure to mediate activation. Here we will use examples from the cell cycle signaling cascade and steroid hormone receptor pathways to highlight some mechanisms of TF activation. A key element to take away from this section is that transcription factor activation is often highly pleiotropic and has many cellular effects. Depending on the cell type and the environmental conditions, different combinations of downstream target genes may be activated or inactivated. Teasing apart these intricacies and the physiological effects that they have within an organism is a major goal of ongoing research.
Cell Cycle Regulation by p53
p53 is one of the most studied proteins in science. To date, over 68,000 papers appear in PubMed containing p53 or TP53 in the title and/or abstract. Originally described as an oncogene (since a mutated, functionally altered form of the protein was first characterized), p53 is now recognized as the most frequently inactivated tumor suppressor in human cancers. It is a transcription factor that controls the expression of genes and miRNAs affecting many important cellular processes including proliferation, DNA repair, programmed cell death (apoptosis), autophagy, metabolism, and cell migration, as shown in Figure \(\PageIndex{1}\). Many of those processes are critical to a variety of human pathologies and conditions extending beyond cancer, including ischemia, neurodegenerative diseases, stem cell renewal, aging, and fertility. Notably, p53 also has non-transcriptional functions, ranging from intrinsic nuclease activity to activation of mitochondrial Bak (Bcl-2 homologous antagonist killer) and caspase-independent apoptosis.
As a transcription factor, p53 responds to various genotoxic insults and cellular stresses (e.g., DNA damage or oncogene activation) by inducing or repressing the expression of over a hundred different genes. p53 transcriptional regulation plays a dominant role in causing the arrest of damaged cells, facilitating their repair and survival, or inducing cell death when DNA is damaged irreparably. p53 can also cause cells to become permanently growth arrested, and there is compelling in vivo evidence that these “senescent” cells secrete factors that enhance their clearance by the immune system, leading to tumor regression. Through these mechanisms, p53 helps maintain genomic stability within an organism, justifying its long-held nickname “guardian of the genome”. Other p53 gene targets are involved in inhibiting tumor cell angiogenesis, migration, metastasis, and other important processes (such as metabolic reprogramming) that normally promote tumor formation and progression

Figure \(\PageIndex{2}\) shows an interactive iCn3D model of the human p53 tetramer bound to the natural CDKN1A(p21) p53-response element (3TS8).
_p53-response_element_(3TS8).png?revision=1&size=bestfit&width=503&height=476)
Each monomer of the p53 tetramer is shown in a different color. The noncovalent interactions of the brown monomer with the DNA are shown, with key amino acids and nucleic acid bases shown in CPK-colored sticks. They interact with the Zn2+ ions, shown in the light green monomer.
Normally, p53 levels are kept low by its major antagonist, Mdm2, an E3 ubiquitin ligase that is itself a transcriptional target of p53. Stress signals, such as DNA damage, oncogene activation, and hypoxia, promote p53 stability and activity by inducing post-translational modifications (PTMs) and tetramerization of p53. p53 functions as a transcription factor that binds to specific p53 response elements upstream of its target genes. p53 affects many important cellular processes linked to tumor suppression, including the induction (green) of senescence, apoptosis, and DNA repair as well as inhibition (red) of metabolism, angiogenesis, and cell migration. These functions are largely mediated through transcriptional regulation of its targets (examples given).
p53 protein function is regulated post-translationally by coordinated interaction with signaling proteins including protein kinases, acetyltransferases, methyl-transferases, and ubiquitin-like modifying enzymes as shown in Figure \(\PageIndex{3}\). The majority of the sites of covalent modification occur at intrinsically unstructured linear peptide docking motifs that flank the DNA-binding domain of p53 which plays a role in anchoring or in allosterically activating the enzymes that mediate covalent modification of p53. In undamaged cells, the p53 protein has a relatively short half-life and is degraded by a ubiquitin-proteasome-dependent pathway through the action of E3 ubiquitin ligases, such as MDM2 (Fig 28.3.1). Following stress, p53 is phosphorylated at multiple residues, thereby modifying its biochemical functions required for increased activity as a transcription factor. Post-translational modifications help to stabilize the tetramer formation of the protein and enhance the translocation of the protein from the cytoplasm into the nucleus. The tetrameric form of p53 is then functional to bind to DNA in a sequence-specific manner and either activate or repress transcription, depending on the target sequence. Some post-translational modifications, such as acetylation, are DNA-dependent and can play a role in chromatin remodeling and activation of p53 target gene expression.

It should be noted that single-point mutations that modify the ability of the protein to be phosphorylated in one position, typically do not show a decrease in the stabilization or activation of the protein following a damage or stress event. Thus, multiple modifications likely allow for redundancy within this pathway and ensure the activation of the protein following a stress event. Furthermore, the environment within the cell can lead to different p53 phenotypes, such as the activation of growth arrest and DNA repair processes (ie if there is not a lot of damage) or it can lead to the activation of apoptosis or programmed cell death pathways (ie if the damage is too extensive to be repaired).
Steroid Hormone Receptors
Steroid hormone receptors (SHRs) belong to the superfamily of nuclear receptors (NRs), which are one of the essential classes of transcriptional factors. NRs play a critical role in all aspects of human development, metabolism. and physiology. Since they generally act as ligand-activated transcription factors, they are an essential component of cell signaling. NRs form an ancient and conserved family that arose early in the metazoan lineage. NR molecular evolution is characterized by major events of gene duplication and gene losses. Phylogenetic analysis revealed a distinct separation of NR ligand binding domains (LBDs) into 4 monophyletic branches, the steroid hormone receptor-like cluster, the thyroid hormone-like receptors cluster, the retinoid X-like and steroidogenic factor-like receptor cluster and the nerve growth factor-like/HNF4 receptor cluster, as shown in Figure \(\PageIndex{4}\).

Here we will focus on the Steroid Hormone-Like Receptors branch (SHRs). SHRs plays a key role in many important physiological processes like organ development, metabolite homeostasis, and response to external stimuli. The estrogen receptor comes in two major forms, ERα and ERβ. Other members of this subgroup include the cortisol-binding glucocorticoid receptor (GR), the aldosterone-binding mineralocorticoid receptor (MR), the progesterone receptor (PR), and the dihydrotestosterone (DHT) binding androgen receptor (AR), as shown in Figure \(\PageIndex{5}\ below.
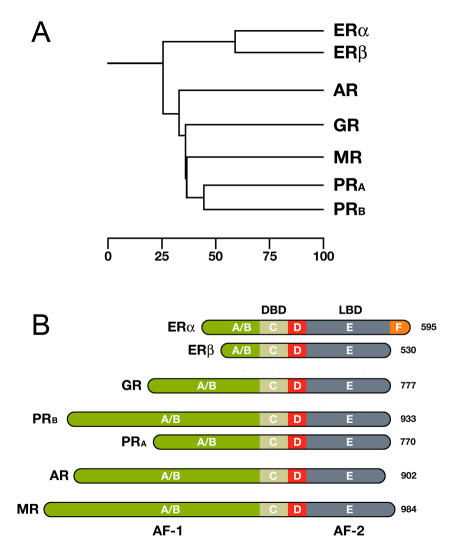
Panel A shows a phylogenetic tree of the Steroid Hormone Receptor (SHR) family showing the evolutionary interrelationships and distance between the various receptors. Based on alignments available at The NucleaRDB [Horn et al., 2001].
Panel B shows that all steroid receptors are composed of a variable N-terminal domain (A/B) containing the AF-1 transactivation region, a highly conserved DNA Binding Domain (DBD), a flexible hinge region (D), and a C-terminal Ligand Binding Domain (LBD, E) containing the AF-2 transactivation region. The estrogen receptor α is unique in that it contains an additional C-terminal F domain. Numbers represent the length of the receptor in amino acids.
The members of the Steroid Hormone Receptor family share a similar, modular architecture, consisting of several independent functional domains (Fig. 5B above). Most conserved is the centrally located DNA binding domain (DBD) containing the characteristic zinc-finger motifs. The DBD is followed by a flexible hinge region and a moderately conserved Ligand Binding Domain (LBD), located at the carboxy-terminal end of the receptor. The estrogen receptor α is unique in that it contains an additional F domain of which the exact function is unclear. The LBD is composed of twelve α-helices (H1-H12) that together fold into a canonical α-helical sandwich. Besides its ligand binding capability, the LBD also plays an important role in nuclear translocation, chaperone binding, receptor dimerization, and coregulator recruitment through its potent ligand-dependent transactivation domain, referred to as AF-2. A second, ligand-independent, transactivation domain is located in the more variable N-terminal part of the receptor, designated as AF-1. To date, no crystal structure of a full-length SHR exists, though structures of the DBD and LBD regions of most SHRs are available. These have helped significantly in understanding the molecular aspects of DNA and ligand binding, but have to some extent also led to biased attention to these parts of the receptor only. For example, many coregulator interaction studies are still performed with the LBD only, while numerous studies have demonstrated that the AF-2 domain often tells only part of the story. With the help of biophysical techniques, however, it is feasible to study the full-length receptor in its native environment.
Most SHRs remain in the cytoplasm of the cell until they are bound with the appropriate steroid as shown in Figure \(\PageIndex{6}\). Steroid binding causes the dimerization of SHRs and localization to the cell nucleus, where the SHRs interact with the DNA at sequence-specific motifs known as Hormone Response Elements (HREs) (Step 5). Many SHRs can also interact with membrane-bound receptors and affect cellular signaling pathways, in addition to the activation of gene expression (step 6).
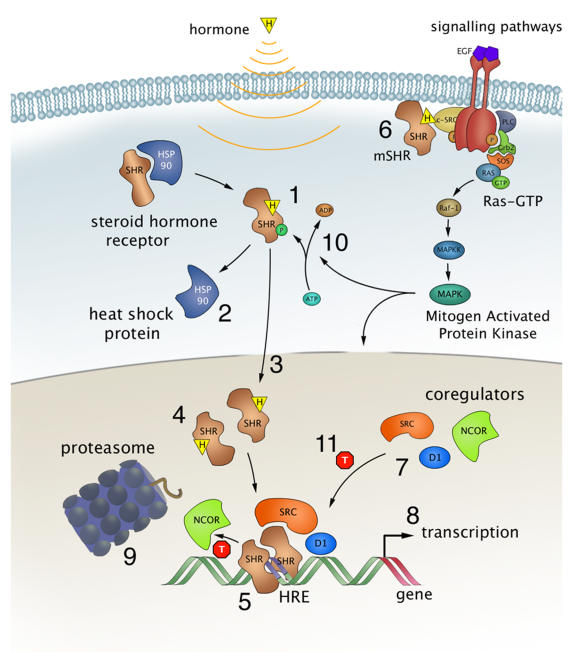
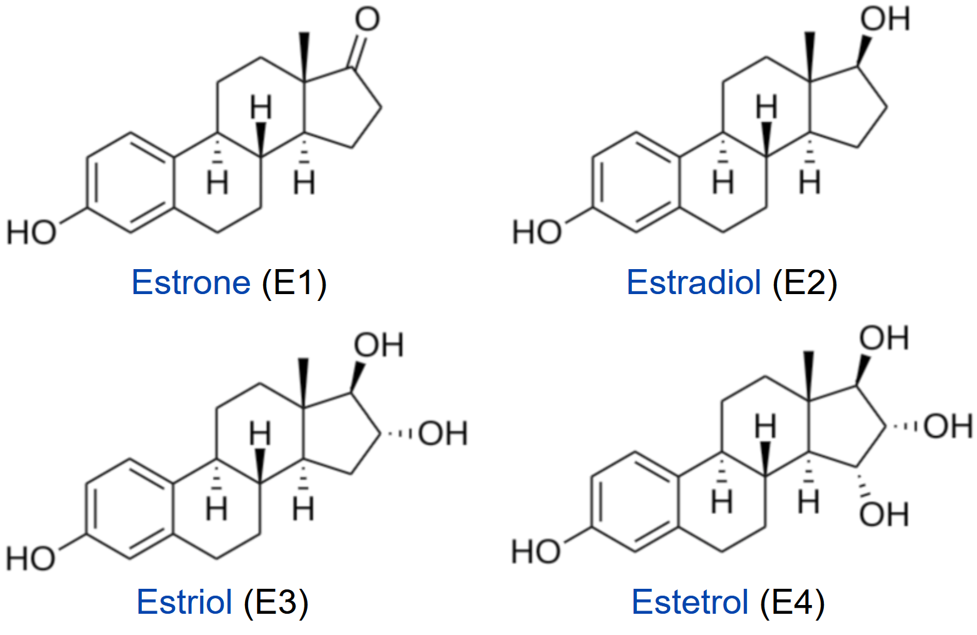
Steroid hormones, such as estrogens, reach their target cells via the blood, where they are bound to carrier proteins. Naturally occurring estrogens including estradiol, estrone, estriol, differ primarily in structure on the presence of hydroxyl groups (Fig. 28.3.6). Estradiol is the predominant estrogen during reproductive years both in terms of absolute serum levels as well as in terms of estrogenic activity. During menopause, estrone is the predominant circulating estrogen, and during pregnancy, estriol is the predominant circulating estrogen in terms of serum levels. Another type of estrogen called estetrol (E4) is produced also produced predominantly during pregnancy as shown in Figure \(\PageIndex{7}\). Estrogens function in many physiological processes, including the regulation of the menstrual cycle and reproduction, maintaining bone density, brain function, cholesterol mobilization, and maturation of reproductive organs during development, and they play a role in controlling inflammation.
Because of their lipophilic nature, it is thought that steroid hormones, such as estrogen, pass the cell membrane by simple diffusion, although some evidence exists that they can also be actively taken up by the endocytosis of carrier protein-bound hormones. For a long time, it has been assumed that binding of the ligand resulted in a simple on/off switch of the receptor (Fig. 6, step 1). While this is likely the case for typical agonists like estrogen and progesterone, this is not always correct for receptor antagonists, used in drug therapy. These antagonists come in two kinds, so-called partial antagonists (for the estrogen receptors known as SERMs for Selective Estrogen Receptor Modulators) and full antagonists. The partial antagonist can, depending on cell type, act as a SHR agonist or antagonist. In contrast, full antagonists (for ER known as SERDs for Selective Estrogen Receptor Downregulators) always inhibit the receptor, independent of cell type, in part by targeting the receptor for degradation. Binding of either type of antagonist results in major conformational changes within the LBD and in the release from heat shock proteins that thus far had protected the unliganded receptor from unfolding and aggregation (Fig. 6 step 2).
Figure \(\PageIndex{8}\) shows an interactive iCn3D model of the Androgen Receptor DNA-Binding Domain Bound to a Direct Repeat Response Element (1R4I).
.png?revision=1&size=bestfit&width=544&height=317)
Transcription Factor (TF) Recognition and Binding to DNA
TF controls gene expression by binding to their target DNA site to recruit, or block, the transcription machinery onto the promoter region of the gene of interest. Their function relies on the ability to find their target site quickly and selectively. In living cells, TFs are present in nM concentrations and bind the target site with comparable affinity, but they also bind any DNA sequence (nonspecific binding), resulting in millions of low affinity (i.e., >10−6 M) competing sites. Nonspecific binding facilitates the search for the target site by three major mechanisms as shown in Figure \(\PageIndex{9}\).
The second scenario is a ‘hopping’ mechanism, in which a TF might hop from one site to another in 3D space by dissociating from its original site and subsequently binding to the new site. This may happen within the same chain and re-association occurs adjacent to the former dissociated site. A third search mechanism is described as ‘intersegmental transfer’. In this scenario, the protein moves between two sites via an intermediate ‘loop’ formed by the DNA and subsequently binds at two different DNA sites. This mechanism applies to TFs with two DNA-binding sites. Proteins with two DNA-binding sites can occasionally bind non-specifically to two locations situated far apart within the DNA strand, that are brought into close contact through the formation of these loops. Such TFs transfer across a point of close contact without dissociating from the DNA.

Top: When the transcription factor (pink ring) moves from one site to another by sliding along the DNA and is transferred from one base pair to another without dissociating from the DNA, this mechanism is called sliding.
Center: Hopping occurs when the transcription factor moves on the DNA by dissociating from one site and re-associating with another site.
Bottom: Intersegmental transfer describes the mechanism by which the transcription factor gets transferred through DNA bending or the formation of a DNA loop, resulting in the protein being bound transiently to both sides and subsequently moving from one site to the other.
One of the main scenarios involves a ‘sliding’ mechanism, in which the protein moves from its initial non-specific site to its actual target site by sliding along the DNA (also known as 1-dimensional (1D) sliding). When the TF starts to move and shift counterions from the phosphate backbone, the same number of counterions binds to the site left free by the protein. The sliding rate is also dependent on the hydrodynamic radius of the protein; the required rotational movement over the DNA backbone is greater for larger proteins, that tend to slide slowly.
Recent Updates: 9/25/23
The sliding model that was proposed by Von Hippel and Berg suggests that DNA-binding protein exists in two interconverting conformations. One is a specific form (O) that can bind to a target DNA sequence, such as an operator in DNA, through specific hydrogen bonds (along with electrostatic interaction) characterized by a low KD. The other is a nonspecific form (D) that binds mainly with weaker affinity through electrostatic interactions and a high KD. Nonspecific binding brings the protein to the DNA surface. Dynamic conformational changes from the O to D allow sampling of hydrogen bonds between donor and acceptors in the protein and in the major grove of the protein. The protein can diffuse much more quickly along the DNA to find its target site since the search for the specific target site is now effectively 1D instead of 3D. There is no thermodynamic barrier to sliding since counterions that leave the DNA when bound to the protein rebind behind it as the protein slides. These processes are illustrated in Figure \(\PageIndex{10}\) below.

Figure \(\PageIndex{10}\): Two-state model sliding model of DNA protein binding to its specific binding site (operator)
There are a large number of overlapping nonspecific binding sites (let's say each is 6 base pairs in length), which also help drive the nonspecific binding of the protein to the DNA through entropy increases (i.e consider the probability of binding to 1 site on the DNA versus multitudes of overlapping sites). Experiments show that the on-rate for a DNA binding protein for finding its target site increases with increasing length of the DNA molecule the specific site is embedded in. The opposite would be expected given the diffusion rate of large molecules. In fact, the kon was found to be greater than diffusion-controlled limits, which can be explained by the reduced dimensionality of the search for the specific site when the protein is loosely bound through nonspecific electrostatic interactions that enable the 1D search.
Each eukaryotic TF controls tens to hundreds of genes scattered throughout the genome, and expressing each gene needs various TFs simultaneously binding to their sites to form the transcription complex, an extremely rare event in probabilistic terms. As a result, the in vivo site occupancy patterns of eukaryotic TFs are more complex than predicted by their in vitro site-specific binding profiles and do not strongly correlate with the actual levels of gene expression. An interesting feature highlighted by genome analysis is an accumulation of potential TF binding sites in regions flanking eukaryotic genes. Such clusters of degenerate recognition sites are assumed to be key for transcription control and thus are generally classified as gene regulatory regions (RR). For example, the affinity of the Drosophila TF Engrailed to the RRs of its target genes is strongly amplified by long tracts of degenerate consensus repeats that are present in such regions.
Role of Short Tandem Repeats (STRs) in the Genome
As we have mentioned previously, only about 1.5% of the human genome encodes genes for actual proteins. Much of the genome is transcribed at low levels into RNAs, some of which have clearly defined functions (examples include rRNA, mRNAs, and regulatory RNAs). A large part is presumably involved in facilitating the 3D organization of the genome and its dynamic architecture, which determines its replicative and transcriptive access. One poorly understood feature of genomic DNA is short tandem repeats (STRs). These repeats stretch up to 100 nucleotides in length with each repetitive tandem repeat running from just 1 to 6 bases long. They comprise about 6% of the genome (compared to the 1.5% for protein-coding genes), and are found in abundance in chromatin that is transcribed mRNA for proteins. For example, a specific DNA sequence might be GTCACGTGAC while a small STR would be (CG)6C(CG)11
Specifically, STRs surround sites where classic transcription factors (TF) bind. As we described in this and the previous chapter sections, TFs bind through specific DNA binding motifs (like helix-loop-helix or Zn2+ fingers) to consensus sequences (such as response elements and enhancers) in the DNA as they function to control transcription. The binding of transcription factors or other proteins to target sequences occurs initially through nonspecific electrostatic interactions, followed by a dimensionally restricted diffusion along the DNA as the protein finds its specific sequence target.
In contrast, the STRs offer little sequence uniqueness for high affinity, low KD binding sites for specific protein interactions, so the question remains as to how they express function, which their omnipresence suggests they have. Studies by Horton et al (Science 381, 1304 (2023) have shown that classic TFs do indeed bind STRs, albeit at lower affinity (higher KD) compared to their binding to classic TF-specific sequences. The bind with higher affinity than to nonspecific DNA sequences. Just as nonspecific interactions facilitate TF binding to promoter sites, so do the multiple STRs that straddle the DNA binding element.
If a TF binds its isolated target DNA with a low effective KD (high affinity) and -ΔG0 value, a target DNA surrounded by multiple STRs would have an even lower effective KD (higher affinity) and even more -ΔG0 value. They do so by increasing the effective on rate (kon) for protein binding. (Remember that KD = koff/kon. ) The effective size of the target for the TF becomes greater when it is an “island” in the middle of a STR “sea”. The increased affinity stems in large part from the more favorable entropy of having the TF bind not to just 1 site but effectively to multiple overlapping sites. This also increases the localized concentration of TF near the specific site which drives binding. The koff is not expected to change.
Another effect of the STRs on TF binding is that multiple DNA-binding proteins can bind to the same site through their interactions with STRs at the site, leading to new ways to regulate gene transcription. The group studies just two TFS but sequence analyses suggest that many TF would use a similar mechanism.
Histone Modification and Chromatin Remodeling
Regulation of transcription involves dynamic rearrangements of chromatin structure. Recall that eukaryotic DNA is complexed with histone octamers, which are composed of dimers of the core histones H2A, H2B, H3, and H4. 147 bp of DNA are wrapped 1.65 times around each octamer forming nucleosomes, the basic packaging units of chromatin. Nucleosomes, connected by linker DNA of variable length as “beads on a string”, generate the 11 nm linear structure. The linker histone H1 is positioned at the top of the core histone octamer and enables higher organized compaction of DNA into transcriptionally inactive 30 nm fibers.
To understand the role of chromatin in the regulation of transcription it is important to know where nucleosomes are positioned and how the positioning is achieved. Basically, there are four groups of activities that change chromatin structure during transcription: (1) histone modifications, (2) eviction and repositioning of histones, (3) chromatin remodeling, and (4) histone variant exchange. Histone modifiers introduce post-translational, covalent modifications to histone tails and thereby change the contact between DNA and histones. These modifications govern access to regulatory factors. Histone chaperones aid in the eviction and positioning of histones. A third class of chromatin restructuring factors is ATP-dependent chromatin remodelers. These multi-subunit complexes utilize energy from ATP hydrolysis for various chromatin remodeling activities including nucleosome sliding, nucleosome displacement, and the incorporation and exchange of histone variants.
Post-translational modifications (PTMs) of histone proteins are a primary mechanism that controls chromatin architecture. Over 20 distinct types of histone PTMs have been described, among which the most abundant ones are acetylation and methylation of lysine residues. Histone PTMs can be deposited on and removed from chromatin by different enzymes, known as histone PTM ‘writers’ and ‘erasers’. Histone PTMs exert their regulatory effects via two main mechanisms. First, histone PTMs serve as docking sites for various nuclear proteins––histone PTM ‘readers’––that specifically recognize modified histone residues through their modification-binding domains. Recruitment of these proteins at specific genomic loci promotes key chromatin processes, such as transcriptional regulation and DNA damage repair. Second, some histone PTMs, such as acetylation, directly affect chromatin's higher-order structure and compaction, thereby controlling chromatin accessibility to protein machinery such as those involved in transcription. Chromatin may adopt one of two major states interchangeably. These states are heterochromatin and euchromatin. Heterochromatin is a compact form that is resistant to the binding of various proteins, such as transcriptional machinery. In contrast, euchromatin is a relaxed form of chromatin that is open to modifications and transcriptional processes, as shown in Figure \(\PageIndex{10}\). Histone methylation promotes the formation of Heterochromatin whereas, histone acetylation promotes euchromatin.

The addition of methyl groups to the tails of histone core proteins leads to histone methylation, which in turn leads to the adoption of a condensed state of chromatin called ‘heterochromatin.’ Heterochromatin blocks transcription machinery from binding to DNA and results in transcriptional repression. The addition of acetyl groups to lysine residues in the N-terminal tails of histones causes histone acetylation, which leads to the adoption of a relaxed state of chromatin called ‘euchromatin.’ In this state, transcription factors and other proteins can bind to their DNA binding sites and proceed with active transcription.
Chromatin remodeling can also be an ATP-dependent process and involve histone dimer ejection, full nucleosome ejection, nucleosome sliding, and histone variant exchange as shown in Figure \(\PageIndex{12}\). ATP-dependent chromatin remodeling complexes bind to nucleosome cores and the surrounding DNA, and, using energy from ATP hydrolysis, they disrupt the DNA-histone interactions, slide or eject nucleosomes, alter nucleosome structures, and modulate the access of transcription factors to the DNA (Figure 28.3.9). In addition to modulating gene expression, some of the complexes are involved in nucleosome assembly and organization, following transcription at locations in which nucleosomes have been ejected, packing of DNA, following replication, and DNA repair.
![Overview of the functions of ATP-dependent chromatin remodeling complexes. (a) A subset of ISWI and CHD complexes are involved in nucleosome assembly, maturation, and spacing. (b) SWI/SNF complexes are primarily involved in histone dimer ejection, nucleosome ejection, and nucleosome repositioning through sliding, thus modulating chromatin access. (c) INO80 complexes are involved in histone exchange. It should be noted that the complexes might be involved in other chromatin remodeling functions (figure adapted from [52]).](https://bio.libretexts.org/@api/deki/files/49680/Overview-of-the-functions-of-ATP-dependent-chromatin-remodeling-complexes-a-A-subset.png?revision=1&size=bestfit&width=588&height=498)
Panel (a) shows a subset of ISWI and CHD complexes is involved in nucleosome assembly, maturation, and spacing.
Panel (b) shows SWI/SNF complexes are primarily involved in histone dimer ejection, nucleosome ejection, and nucleosome repositioning through sliding, thus modulating chromatin access.
Panel (c) shows INO80 complexes are involved in histone exchange. It should be noted that the complexes might be involved in other chromatin remodeling functions.
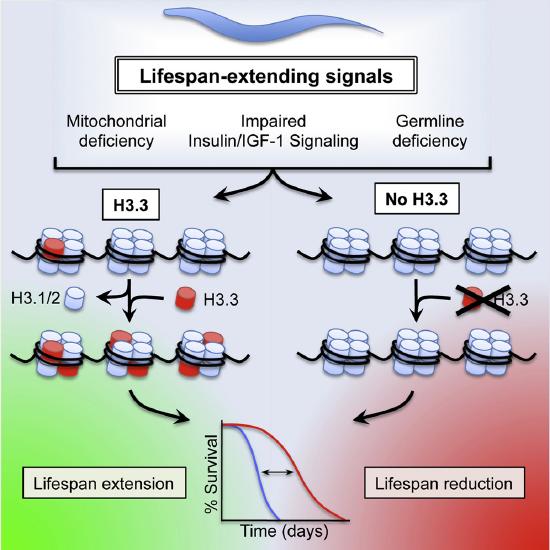
Figure \(\PageIndex{13}\)s shows the effects of Histone Variant H3.3 on C. elegans Lifespan
Protein-DNA Interactions
Proteins use a wide range of DNA-binding structural motifs, such as homeodomain (HD), helix-turn-helix (HTH), and high-mobility group box (HMG) to recognize DNA. HTH is the most common binding motif and can be found in several repressor and activator proteins, as shown in Figure \(\PageIndex{14}\). Despite their structural diversity, these domains participate in a variety of functions that include acting as substrate interaction mediators, enzymes to operate DNA, and transcriptional regulators. Several proteins also contain flexible segments outside the DNA-binding domain to facilitate specific and non-specific interactions. For example, many HD proteins use N-terminal arms and a linker region to interact with DNA. The Encyclopedia of DNA Elements (ENCODE) data suggest that about 99.8% of putative binding motifs of TFs are not bound by their respective TFs in the genome. It is, therefore, clear that the presence of a single binding motif per TF is not adequate for TF binding.

Figure \(\PageIndex{14}\): Representative figures of the transcription factor binding domains. The figure shows the crystal structures of different types of TF domains (3l1p, 4m9e, 5d5v, 1lbg, 1gt0, and 1nkp). The structures were obtained from the Protein Data Bank (PDB) and redrawn using chimera. The respective domains and important regions have been labeled. HTH stands for the helix-turn-helix domain. bHLH stands for the basic helix-loop-helix motif. HD and HMG stand for homeodomain and high-mobility group box domain, respectively. Yesudhas, D., et.al. (2017) Genes 8(8):192
Figure \(\PageIndex{14}\) shows interactive iCn3D models of the transcription factor binding domains as depicted in the figure above. (Copyright; author via source)
|
POU protein:DNA complex HTH-HD domain (3l1p)
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...fHmWMvUCiJus77 |
Human Hsf1 with Satellite III repeat DNA - HTH Domain (5d5v)
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...qkHMwpH7LdDmr7 |
POU-HMG-DNA ternary complex - HTM-HMG domain (1gt0)
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...b9X7DEGSK2n2V7 |
|
Klf4 zinc finger DNA binding domain in complex with methylated DNA(4m9e)
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...T7ZXxMhQ66WNL9 |
Lactose operon repressor and its complexes with DNA and inducer (1lbg)
Click the image for a popup or use this external link:https://structure.ncbi.nlm.nih.gov/i...6v3y24U7N2qYC8 |
Myc-Max and Mad-Max recognizing DNA(1nkp)
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...qcTBN4UwxPFH99 |
.png?revision=1)
.png?revision=1&size=bestfit&width=178&height=232)
.png?revision=1)
.png?revision=1&size=bestfit&width=253&height=297)
.png?revision=1&size=bestfit&width=214&height=297)
.png?revision=1&size=bestfit&width=243&height=301)
Most of the search mechanistic studies that try to determine how TFs find their binding sites are limited to naked DNA-protein complexes, which do not reflect the actual crowded environment of a cell. Studies with naked DNA and transcription factors have shown that many DNA-binding proteins travel a long distance by 1D diffusion. However, the search process for eukaryotes must occur in the presence of chromatin, which can hinder protein mobility. In this case, the protein must dissociate from the DNA, enter a 3D mode of diffusion state, and continue the target site searching process. The sliding and intersegmental transfer mechanisms can be explained through the example of the lac repressor. The lac repressor contains 4 identical monomers (a dimer of dimers) for its DNA binding. The binding sequence of these dimers is symmetric or pseudo-symmetric, and each half is identified by these identical monomers. The HTH domain of the lac repressor is the DNA-binding domain that facilitates the interaction with its target site on DNA as shown in Figure \(\PageIndex{15}\).
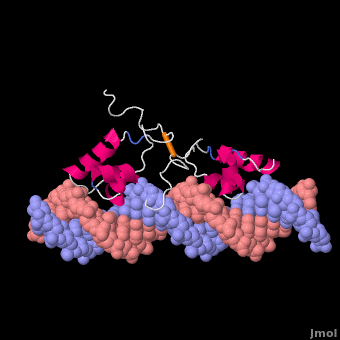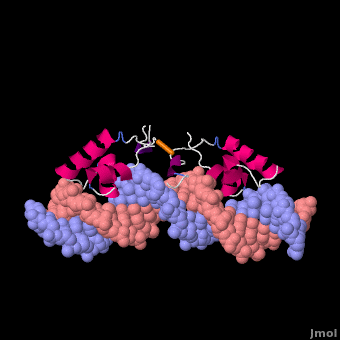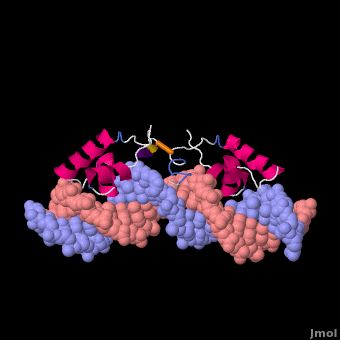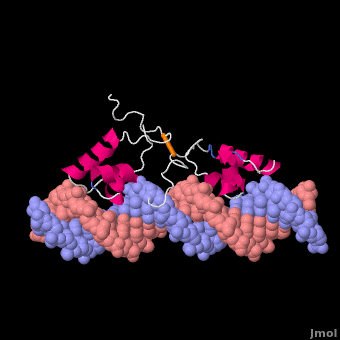
As a result of a rapid search (sliding) along the DNA molecule and intersegmental transfer between distant DNA sequences, the lactose repressor finds its target sites faster than the diffusion limit. The section comprised between residues 1–46 of the HTH protein domain, characterized by three α-helices, maintains its secondary structure through specific and non-specific binding. When the repressor binds to a non-specific site, the HTH domain interacts with the DNA backbone and maintains the interaction with its helix region in the major groove juxtaposition. This arrangement facilitates the interaction of the recognition helix with the edges of the DNA bases, enabling the repressor to walk or search for its specific site on the DNA. The C-terminal residues of the DNA-binding domain, residues 47–62, form the hinge region, and are normally disordered during non-specific recognition; however, during specific site recognition, residues 50–58 acquire an α-helix configuration (hinge helix) (Fig. 15 above). The disordered hinge region and the flexibility of the HTH domain allow the protein to move freely along the DNA to search for its target site. In specific binding complexes, the hinge helix of each monomer is located at the symmetrical center of the binding site, thereby causing the hinge helices to interact with each other (intersegmental transfer) to allow better stability. Moreover, DNA bends at the symmetrical center of the specific binding site (37° angle), thereby supporting monomer-monomer interactions.
In addition to the helix-turn-helix structure, the zinc finger motif is also very common, especially in eukaryotic TFs, as shown in Figure \(\PageIndex{16}\). Proteins that contain zinc fingers (zinc finger proteins) are classified into several different structural families. Unlike many other clearly defined supersecondary structures such as Greek keys or β hairpins, there are a number of types of zinc fingers, each with a unique three-dimensional architecture. A particular zinc finger protein’s class is determined by this three-dimensional structure, but it can also be recognized based on the primary structure of the protein or the identity of the ligands coordinating the zinc ion. Despite the large variety of these proteins, however, the vast majority typically function as interaction modules that bind DNA, RNA, proteins, or other small, useful molecules. Variations in structures serve primarily to alter the binding specificity of a particular protein. The most common type of zinc finger motif utilizes two Cys and two His residues (CCHH) coordinating the Zn(II) ion to adopt a ββα fold with three hydrophobic residues responsible for the formation of a small hydrophobic core which offers additional stabilization of the zinc finger domain.

Panel (a) shows the alignment of the TFIIIA-like zinc finger domains from different organisms. The green color denotes residues that are responsible for the hydrophobic core formation in most CCHH zinc fingers (L17, F11, and L2). Yellow and blue indicate the coordinating Cys and His residues, respectively.
Panel (b) shows the 3D NMR structure of 15-th ZF from zinc finger protein 478 [PDB: 2YRH]
Figure \(\PageIndex{17}\) shows an interactive iCn3D model of C2H2-type zinc finger domain (699-729) from zinc finger protein 473 (2YRH).
_from_zinc_finger_protein_473_(2YRH).png?revision=1&size=bestfit&width=730&height=237)
Overall, zinc finger motifs display considerable versatility in binding modes, even between members of the same class (e.g., some bind DNA, others protein), suggesting that they are stable scaffolds that have evolved specialized functions. For example, zinc finger-containing proteins function in gene transcription, translation, mRNA trafficking, cytoskeleton organization, epithelial development, cell adhesion, protein folding, chromatin remodeling, and zinc sensing, to name but a few. Zinc-binding motifs are stable structures, and they rarely undergo conformational changes upon binding their target.
The last binding domain that we will consider in detail here is the helix-loop-helix domain found in Leucine zipper-containing proteins. Specifically, bZIPs (Basic-region leucine zippers) are a class of eukaryotic transcription factors. The bZIP domain is 60 to 80 amino acids in length with a highly conserved DNA binding basic region and a more diversified leucine zipper dimerization region. The two regions form α-helical structures that are connected via a looped region. This forms a core helix-loop-helix (HLH) structure within each monomer of the protein. Two monomers then join through the formation of a leucine zipper junction forming a heterodimeric protein structure. The resulting heterodimer can bind with DNA in a sequence-specific manner through the basic α-helices as shown in Figure \(\PageIndex{18}\).
.svg?revision=1&size=bestfit&width=410&height=313)
Specifically, basic residues, such as lysines and arginines, interact in the major groove of the DNA, forming sequence-specific interactions ). Most bZIP proteins show a high binding affinity for the ACGT motifs. The bZIP heterodimers exist in a variety of eukaryotes and are more common in organisms with higher evolution complexity.
Figure \(\PageIndex{19}\) shows an interactive iCn3D model of the GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted alpha helices (1YSA).
.png?revision=1&size=bestfit&width=446&height=323)
Epigenetics and Transgenerational Inheritance
Even though all somatic cells of a multicellular organism have the same genome, different cell types have different transcriptomes (sets of all expressed RNA molecules), different proteomes (sets of all proteins), and, hence, different functions. Cell differentiation during embryonic development requires the activation and repression of specific sets of genes by the action of cell lineage-defining transcription factors. Within a cell lineage, gene activity states are often maintained over several rounds of cell divisions (a phenomenon called “cellular memory” or “cellular inheritance”). Since the rediscovery of epigenetics some 30 years ago (it was originally proposed by Conrad Hal Waddington in the early 1940s), cellular inheritance has been attributed to gene regulatory feedback loops, chromatin modifications (DNA methylation and histone modifications) as well as long-lived non-coding RNA molecules, which collectively are called the “epigenome”. Among the different chromatin modifications, DNA methylation and polycomb-mediated silencing are probably the most stable ones and endow genomes with the ability to impose silencing of transcription of specific sequences even in the presence of all of the factors required for their expression.
Defining Transgenerational Epigenetic Inheritance
The metastability of the epigenome explains why development is both plastic and canalized, as originally proposed by Waddington. Although epigenetics deal only with the cellular inheritance of chromatin and gene expression states, it has been proposed that epigenetic features could also be transmitted through the germline and persist in subsequent generations. The widespread interest in “transgenerational epigenetic inheritance” is nourished by the hope that epigenetic mechanisms might provide a basis for the inheritance of acquired traits. Yes, Lamarck has never been dead and every so often raises his head, this time with the help of epigenetics.
Although acquired traits concerning body or brain functions can be written down in the epigenome of a cell, they cannot easily be transmitted from one generation to the next. For this to occur, these epigenetic changes would have to manifest in the germ cells as well, which in mammals are separated from somatic cells by the so-called Weismann barrier. Further, the chromatin is extensively reshaped during germ cell differentiation as well as during the development of totipotent cells after fertilization, even though some loci appear to escape epigenetic reprogramming in the germline. Long-lived RNA molecules appear to be less affected by these barriers and therefore more likely to carry epigenetic information across generations, although the mechanisms are largely unsolved.
Evidence for Transgenerational Epigenetic Inheritance
In the past 10 years, numerous reports on transgenerational responses to environmental or metabolic factors in mice and rats have been published. The factors include endocrine disruptors, high-fat diet, obesity, diabetes, undernourishment as well as trauma. These studies investigated DNA methylation, sperm RNA, or both. For example, when male mice are made prediabetic by treatment with streptozotocin it affects the DNA methylation patterns in their resulting sperm, as well as the pancreatic islets of F1 and F2 of the resulting offspring. Furthermore, studies have shown that traumatic stress in early life altered behavioral and metabolic processes in the progeny and that injection of sperm RNAs from traumatized males into fertilized wild-type oocytes reproduced the alterations in the resulting offspring.
In humans, epidemiological studies have linked food supply in the grandparental generation to health outcomes in the grandchildren. An indirect study based on DNA methylation and polymorphism analyses has suggested that sporadic imprinting defects in Prader–Willi syndrome are due to the inheritance of a grandmaternal methylation imprint through the male germline. Because of the uniqueness of these human cohorts, these findings still await independent replication. Most cases of segregation of abnormal DNA methylation patterns in families with rare diseases, however, turned out to be caused by an underlying genetic variant. Thus, studies of this nature must rule out the effects of traditional genetic inheritance as being a factor of the observed phenotypes.
Genetic inheritance alone cannot fully explain why we resemble our parents. In addition to genes, we inherited from our parents the environment and culture, which in parts have been constructed by the previous generations as shown in Figure \(\PageIndex{20}\). A specific form of the environment is our mother’s womb, to which we were exposed during the first 9 months of our life. The maternal environment can have long-lasting effects on our health. In the Dutch hunger winter, for example, severe undernourishment affected pregnant women, their unborn offspring, and the offspring’s fetal germ cells. The increased incidence of cardiovascular and metabolic disease observed in F1 adults is not due to the transmission of epigenetic information through the maternal germline, but a direct consequence of the exposure in utero, a phenomenon called “fetal programming” or—if fetal germ cells and F2 offspring are affected—“intergenerational inheritance”.
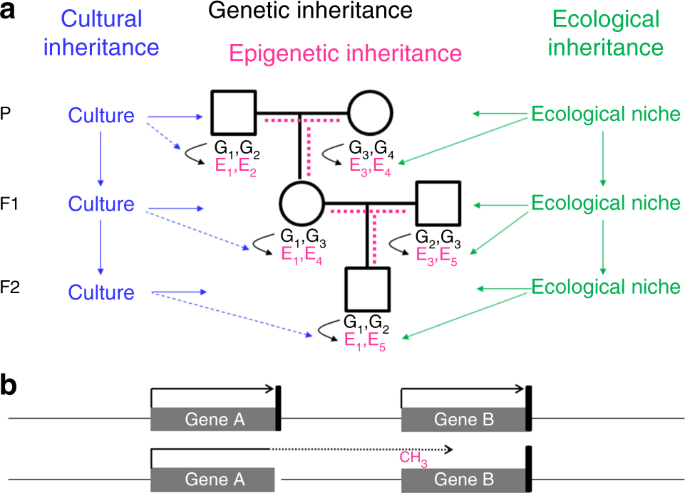
Panel a shows that offspring inherit from their parent's genes (black), the environment (green), and culture (blue). Genes and the environment affect the epigenome (magenta) and the phenotype. Culture also affects the phenotype, but at present, there is no evidence of a direct effect of culture on the epigenome (broken blue lines). It is a matter of debate, how much epigenetic information is inherited through the germline (broken magenta lines). G genetic variant, E epigenetic variant.
Panel b shows that an epimutation (promoter methylation and silencing of gene B in this example) often results from aberrant read-through transcription from a mutant neighboring gene, either in sense orientation as shown here or in antisense orientation. The presence of such a secondary epimutation in several generations of a family mimics transgenerational epigenetic inheritance, although it represents genetic inheritance. Black arrow, transcription; black vertical bar, transcription termination signal; broken arrow, read-through transcription
Roadmap to Proving Transgenerational Epigenetic Inheritance
Here are some steps to show that inheritance is determined by epigenetics and not classical genetics.
- Rule out genetic, ecological, and cultural inheritance. For studies in mice and rats, inbred strains and strictly controlled environments need to be used. When a pregnant female animal is exposed to a specific environmental stimulus, F3 offspring and subsequent generations must be studied to exclude a direct effect of the stimulus on the embryos’ somatic cells and germ cells. Even more desirable is the use of in vitro fertilization (IVF), embryo transfer, and foster mothers. When a male animal is exposed to an environmental stimulus, F2 offspring must be studied to exclude transient effects on germ cells. To ensure that any phenotype is exclusively transmitted via gametes, IVF must be used, controlling for possible artifacts relating to IVF. In contrast with laboratory animals, it is impossible to rule out ecological and cultural inheritance in humans, but genetic effects should and can be excluded. If an epimutation follows Mendelian inheritance patterns, be cautious: you are more likely looking at a secondary epimutation and genetic inheritance. Study the haplotype background of the epimutation: if in a given family it is always on the same haplotype, you are again most likely dealing with a secondary epimutation. Do whole genome sequencing to search for a genetic variant that might have caused the epimutation and be aware that this variant might be distantly located. Good spots to start looking at are the two neighboring genes, where a mutation might cause transcriptional read-through in sense or antisense orientation into the locus under investigation. Unfortunately, if you don’t find anything, you still cannot be 100% sure that a genetic variant does not exist.
- Identify the responsible epigenetic factor in the germ cells. Admittedly, this is easier said than done, especially in female germ cells, which are scarce or unavailable. Be aware that germ cell preparations may be contaminated with somatic cells or somatic DNA. Use swim-up (sperm) or micromanipulation techniques to purify germ cells to the highest purity. Exclude the presence of somatic cells and somatic DNA by molecular testing, for example by methylation analysis of imprinted genes, which are fully methylated or fully unmethylated only in germ cells.
- Demonstrate that the epigenetic factor in the germ cells is responsible for the phenotypic effect in the next generation. If possible, remove the factor from the affected germ cells and demonstrate that the effect is lost. Add the factor to control germ cells and demonstrate that the effect is gained. While RNA molecules can and have been extracted from the sperm of exposed animals and injected into control zygotes, DNA methylation, and histone modifications cannot easily be manipulated (although CRISPR/Cas9-based epigenome editors are being developed and used for this purpose), and all of these experiments can hardly be done in humans. In light of these problems, this might currently be too much to ask for to prove transgenerational epigenetic inheritance in humans, but should, nevertheless, be kept in mind and discussed.
References
- Parker, N., Schneegurt, M., Thi Tu, A-H., Lister, P., Forster, B.M. (2019) Microbiology. Openstax. Available at: https://opentextbc.ca/microbiologyopenstax/
- Chan, K-G., Liu, Y-C., and Chang C-Y. (2015) Inhibiting N-acyl-homoserine lactone synthesis and quenching Pseudomonas quinolone quorum sensing to attenuate virulence. Front. Microbiol. 6:1173. Available at: https://www.frontiersin.org/articles/10.3389/fmicb.2015.01173/full
- Rukavina, Z., and Vanic Zeljka. (2016) Current trends in the development of liposomes targeting bacterial biofilms. Pharmaceutics 8(2):18. Available at: https://www.mdpi.com/1999-4923/8/2/18/htm
- Wikipedia contributors. (2020, April 18). Guanosine pentaphosphate. In Wikipedia, The Free Encyclopedia. Retrieved 16:26, August 23, 2020, from https://en.Wikipedia.org/w/index.php?title=Guanosine_pentaphosphate&oldid=951778776
- Verbeke, F., De Craemer, S., Debunne, N., Janssens, Y., Wynendaele, E., Van de Wiele, C., and De Spiegeleer. B. (2017) Peptides as quorum sensing molecules: measurement techniques and obtained levels in vitro and in vivo. Frontiers in Neuroscience 11:183. Available at: https://www.researchgate.net/publication/316055402_Peptides_as_Quorum_Sensing_Molecules_Measurement_Techniques_and_Obtained_Levels_In_vitro_and_In_vivo
- Yesudhas, D., Batool, M., Anwar, M.A., Panneerselvam, S., and Choi, S. (2017) Proteins recognizing DNA: Structural uniqueness and versatility of DNA-binding domains in Stem Cell Transcription Factors. Genes 8(8):192. Available at: https://www.mdpi.com/2073-4425/8/8/192/htm
- Castellanos, M., Mothi, N., and Muñoz, V. (2020) Eukaryotic transcription factors can track and control their target genes using DNA antennas. Nature Comm. 11:540. Available at: https://www.nature.com/articles/s41467-019-14217-8
- Neideracher, G., Klopf, E., and Schüller, C. (2011) Interplay of dynamic transcription and chromatin remodeling: Lessons from yeast. Int J Mol Sci 12(8):4758-4769. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3179130/
- Kim, S., and Kaang, B-K. (2017) Epigenetic regulation and chromatin remodeling in learning and memory. Exp. & Mol. Med. 49:e281. Available at: https://www.nature.com/articles/emm2016140#Fig1
- Tvardovskly, A., Schwämmle, V., Kempf, S., Rogowska-Wrzesinka, A., and Jensen, O.N. (2016) Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nuc. Acids Res. 45(16):1093. Available at: https://www.researchgate.net/publication/318987684_Accumulation_of_histone_variant_H33_with_age_is_associated_with_profound_changes_in_the_histone_methylation_landscape
- Cipolletti, M., Fernandez, V.S., Montalesi, E., Marino, M., Fiochetti, M. (2018) Beyond the antioxidant activity of dietary polyphenols in cancer: The modulation of estrogen receptors (ERs) signaling. Int J. Mol Sci 19(9)2624. Available at: https://www.mdpi.com/1422-0067/19/9/2624/htm
- Griekspoor, A., Zward, W., Neefjes, J., and Michalides, R. (2007) Visualizing the action of steroid hormone receptors in living cells. Nucl. Recept. Signal. 5:e003 Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1853070/
- Mitsis, T., Papargeorgiou, L., Efthimiadou, A., Bacopoulou, F., Vlachakis, D., Chrousos, G.P., Eliopoulos, E. (2020) A comprehensive structural and functional analysis of the ligand binding domain of the nuclear receptor superfamily reveals highly conserved signaling motifs and two distinct canonical forms through evolution. World Acad Sci J 1: 264-274, 2019. Available at: https://www.spandidos-publications.com/10.3892/wasj.2020.30
- Reed, S.M., and Quelle, D.E. (2015) p53 Acetylation: Regulation and consequences. Cancers 7(1):30-69. Available at: https://www.mdpi.com/2072-6694/7/1/30/htm.
- Maclaine, N.J., and Hupp, T.R. (2009) The regulation of p53 by phosphorylation: a model for how distinct signals integrate into the p53 pathway. Aging 1(5):490-502. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2806026/
- Wikipedia contributors. (2020, August 1). Estrogen. In Wikipedia, The Free Encyclopedia. Retrieved 01:28, September 6, 2020, from https://en.Wikipedia.org/w/index.php?title=Estrogen&oldid=970560042
- Kluska, K., Adamczyk, J., and Krezel, A. (2018) Metal binding properties, stability, and reactivity of zinc fingers. Coord. Chem Rev. 367:18-64. Available at: https://www.sciencedirect.com/science/article/pii/S0010854517305441
- Wikipedia contributors. (2020, July 4). Leucine zipper. In Wikipedia, The Free Encyclopedia. Retrieved 07:00, September 7, 2020, from https://en.Wikipedia.org/w/index.php?title=Leucine_zipper&oldid=965962667
- Wikipedia contributors. (2020, April 15). Zinc finger. In Wikipedia, The Free Encyclopedia. Retrieved 18:28, September 7, 2020, from https://en.Wikipedia.org/w/index.php?title=Zinc_finger&oldid=951116840
- Horsthemke, B. (2018) A critical view on transgenrational epigenetic inheritence in humans. Nat. Comm. 9:2973. Available at: https://www.nature.com/articles/s41467-018-05445-5#rightslink