
22.4: Biosynthesis and Degradation of Nucleotides
- Page ID
- 15181


Introduction
We conclude our exploration of metabolic pathways with the biosynthesis and breakdown of nucleotides, the monomers that comprise nucleic acids. We can't also forget the important role of ATP as the universal carrier of biological free energy, as well as the nucleotides involved in signal transduction (GTP in heterotrimeric G proteins, small G proteins, and ATP as substrate in protein phosphorylations by kinases). As with the other later sections on metabolism, we won't focus much on detailed reaction mechanisms or enzyme structures, with one exception, the enzyme that converts nucleotides to deoxynucleotides.
Nucleotide synthesis is often included in chapters on amino acid metabolism as almost every atom in the purine and pyrimidine ring derives from them as shown in Figure \(\PageIndex{1}\).

Figure \(\PageIndex{1}\): Source of atoms in nucleotide bases. Lieu, E.L., Nguyen, T., Rhyne, S., et al. Amino acids in cancer. Exp Mol Med 52, 15–30 (2020). https://doi.org/10.1038/s12276-020-0375-3. Creative Commons Attribution 4.0 International License, http://creativecommons.org/licenses/by/4.0/.
For purines, glutamine and aspartate provide the nitrogen for the nucleotide's rings. They also provide the NH3s for the ring substituents (glutamine for adenine and aspartate for guanine). Glycine and formate provide the carbon atoms for the rings. The one carbon molecule formate, which derives from glycine, is also added to purine rings. Glycine provides a one-carbon unit indirectly through the main carrier of activated one-carbon units, 5,10-meTHF, which is converted to formate through 10-formyl THF.
Pyrimidines are much smaller and their synthetic pathway reflects that. Instead of being synthesized as nucleobases as in the case of purines, they are made as ribonucleotides as they are linked to phosphoribosyl pyrophosphate (PRPP). Glutamine and aspartate again provide the ring C and N atoms. The one-carbon unit derives from serine-to-glycine conversion. A methyl group from the activated 1C donor, 5,10-meTHF, is added to dUMP to make dTMP.
Purine Synthesis
The material below derives from De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. https://doi.org/10.3390/cells10102603. Creative Commons Attribution License
Mammals have two pathways for purine synthesis, a de novo pathway and a salvage pathway to recycle nucleotide bases. The salvage pathway is typically sufficient as purine bases come from nucleic acid breakdown. The resulting free bases (adenine, guanine, and hypoxanthine are connected to phosphoribosyl pyrophosphate (PRPP) to form nucleoside monophosphates (NMP) using either adenine phosphoribosyltransferase (APRT) to form AMP or hypoxanthine-guanine phosphoribosyltransferase (HGRT) to form IMP and GMP. PRPP is a substrate in both the salvage and de novo pathways. The overall de novo and salvage pathways for purine synthesis are described in detail in Figure \(\PageIndex{2}\).

Figure \(\PageIndex{2}\): Purine metabolic pathways. De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. https://doi.org/10.3390/cells10102603. Creative Commons Attribution License
The conserved de novo biosynthesis pathway to generate IMP consists of 10 chemical steps catalyzed by 6 gene products in humans. These include the trifunctional enzyme TGART, composed of GAR synthetase (GARS), GAR transformylase (GARTfase), and AIR synthetase (AIRS) domains; the bifunctional enzymes PAICS, composed of CAIR synthetase/AIR carboxylase (CAIRS) and SAICAR synthetase (SAICARS), and ATIC, composed of AICAR transformylase (AICART) and IMP cyclohydrolase (IMPCH); and three monofunctional enzymes, phosphoribosyl amidotransferase (PPAT), formyl glycin amidine ribonucleotide synthetase (FGAMS), and adenylosuccinate lyase (ADSL). Downstream IMP is converted to (1) GMP through stepwise reactions of IMP dehydrogenase (IMPDH) followed by GMP synthetase (GMPS) and (2) AMP via adenylosuccinate synthetase (ADSS) followed by ADSL. The salvage pathway requires PRPP to generate IMP and GMP through one-step reactions mediated by hypoxanthine phosphoribosyltransferase (HPRT) utilizing hypoxanthine and guanine bases. AMP is generated by adenine phosphoribosyltransferase (APRT) utilizing adenine base and PRPP as substrates. Mitochondria supply precursors for purine de novo biosynthesis including glycine, N10-formyl THF, and aspartic acid through their one-carbon cycle (1C cycle) and tricarboxylic acid cycle (TCA).
The de novo pathway kicks in when there is high demand for purines. Six enzymes are required for the 10-step pathway. Three of these are multifunctional enzymes catalyzing multiple steps in the pathway, comprising the two bifunctional enzymes phosphoribosylaminoimidazole carboxylase (PAICS) and AICAR transformylase/inosine monophosphate cyclohydrolase (ATIC) and the trifunctional enzyme glycinamide ribonucleotide transformylase (TGART). When active, the pathway is limited both by substrate availability and by the reaction rate of its initial step, the conversion of PRPP to phosphoribosylamine (PRA) by phosphoribosylpyrophosphate amidotransferase (PPAT). The final product of the de novo biosynthesis pathway, IMP is a substrate for the production of both AMP and GMP. 6 ATP are used to make 1 IMP from PRPP. None are required in the salvage pathway.
PPAT is also called Glutamine phosphoribosylpyrophosphate amidotransferase or amidophosphoribosyltransferase. It catalyzes the rate limiting step, is tightly regulated. PPAT possesses two nucleotide-binding sites near the active site, allowing for feedback control by downstream purine nucleotides via allosteric inhibition.
Figure \(\PageIndex{3}\) shows an interactive iCn3D model of the Glutamine Phosphoribosylpyrophosphate Amidotransferase from Arabidopsis thaliana (6LBP)
.png?revision=1&size=bestfit&width=248&height=281)
 Figure \(\PageIndex{3}\): Glutamine Phosphoribosylpyrophosphate Amidotransferase from Arabidopsis thaliana (6LBP). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...uWnf1bTq1odk88
Figure \(\PageIndex{3}\): Glutamine Phosphoribosylpyrophosphate Amidotransferase from Arabidopsis thaliana (6LBP). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...uWnf1bTq1odk88
The plant enzyme is a homotetramer, which each subunit having a Fe4S4 center (spacefill). The active site residues in each subunit (DDS 432-444 and DS 369-370) are shown as colored sticks and labeled.
Regulation of IMP production also occurs through enzyme phosphorylation. For example, Thr 397 on PPAT is phosphorylated by protein kinase B (PKB). PRPP concentrations also affects the rate. Another regulation of the flux through the de novo pathway is through the condensation of the enzymes into the purinosome, which contains PPAT, TGART, formylglycinamidine ribonucleotide synthetase (FGAMS), PAICS, adenylosuccinate lyase (ADSL), and ATIC. The purinosome also interacts with the mitochondria which would allow high local concentrations of ATP.
A cartoon view of the purinosome is shown in Figure \(\PageIndex{4}\).
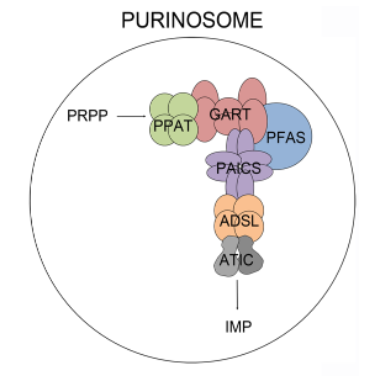
Figure \(\PageIndex{4}\): Cartoon showing the purinosome. Baresova et al (2018) PLoS ONE 13(7): e0201432. https://doi.org/10.1371/journal.pone.0201432. Creative Commons Attribution License,
Figure \(\PageIndex{5}\) shows another view of de novo IMP synthesis in which the origin of each atom in the purine ring is shown in color.

Figure \(\PageIndex{5}\): De novo IMP synthesis showing the origin of atoms in IMP.
Figure \(\PageIndex{6}\) shows an expanded view of the conversion of IMP to GTP and ATP.

Figure \(\PageIndex{6}\): Conversion of IMP to GTP and ATP
Pyrimidine Synthesis
As mentioned in the introduction, pyrimidines have a much simpler biosynthetic pathway. Instead of being synthesized as nucleobases as in the case for purines, they are made as ribonucleotides as they are linked to phosphoribosyl pyrophosphate (PRPP). Glutamine and aspartate again provide the ring C and N atoms. The one-carbon unit derives from serine-to-glycine conversion. A methyl group from the activated 1C donor, 5,10-meTHF, is added to dUMP to make dTMP.
The first step in the pathway for pyrimidine synthesis is the condensation of aspartate and carbamoyl phosphate. We have seen the synthesis of carbamoyl phosphate in the urea cycle by the enzyme carbamoylphosphate synthase I (CPSI) in Chapter 18.3 but present the reaction again in Figure \(\PageIndex{7}\).

Figure \(\PageIndex{7}\): Synthesis of carbamoyl phosphate
A different cytosolic version of the enzyme, CPS II, is used to synthesize both arginine and pyrimidine nucleotides. It uses glutamine as a donor of NH3.
The pathway for the synthesis of UTP and CTP are shown in Figure \(\PageIndex{7}\). It does not explicitly show the synthesis of carbamoylphosphate, which is an intergyral part of the pathway and one of the rate-limiting steps in pyrimidine synthesis.

Figure \(\PageIndex{8}\): De novo synthesis of UDP, UTP and CTP
UDP and CDP can be converted to dCDP and dUDP, then on to dCPT and dUTP, and to dTMP as shown in Figure \(\PageIndex{9}\).

Figure \(\PageIndex{9}\): Synthesis of dTMP
Some of the material below derives from Li et al. Int. J. Mol. Sci. 2021, 22(19), 10253; https://doi.org/10.3390/ijms221910253. Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/)
In purine synthesis, we saw multifunctional enzymes that catalyze several steps, as well as the assembly of the enzymes in the de novo pathway into the purinosome. In an analogous fashion, three different enzyme activities that catalyze the first three combined rate limiting steps of pyrimidine synthesis, Carbamoyl-phosphate synthetase, Aspartate transcarbamoylase, and Dihydroorotase are found in a single, multifunction protein referred to as CAD. Its structure is a hexamer of a 243K monomer. It has 4 domains that include
- glutamine amidotransferase (GATase) which "moves" HCO3−, glutamine, and ATP to the CPSIIase domain
- carbamoylphosphate synthetase II (CPSIIase): This has two parts, CPSaseA, and CPSase B. They combine functionally with GATase to form a glutamine-dependent carbamoylphosphate synthase (CPSase)
- aspartate transcarbamylase (ATCase) acts as a homotrimer
- dihydroorotase (DHOase) catalyzes the reversible cyclization reaction.
The CAD protein is a 'fusion' protein encoding these four enzymatic activities of the pyrimidine pathway. Figure \(\PageIndex{10}\) shows the domain structure of the CAD protein.

Figure \(\PageIndex{10}\): Domain structure of CAD
The red represents glutamine amidotransferase and the blue the carbamoyl phosphate synthase ATP binding domain. More specifically, the following amino acid stretches comprise the different domains: GATase (2-365), CPSase A (395-933), CPSlase B (934-1455), DHOase (1456-1788), and ATCase (1918-2225)
Figure \(\PageIndex{11}\) shows an interactive iCn3D model of the AlphaFold predicted model of the CAD protein (P27708)
.png?revision=1&size=bestfit&width=387&height=262)
Figure \(\PageIndex{11}\): AlphaFold predicted model of the CAD protein (P27708). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...5V2XUmfsz1qjZ6
The GATase domain is magenta, the CPSase domains orange, the DHOase domain yellow, and the ATCase domain cyan. The structure of the disordered loops could not be modeled.
Ribonucleotide reductases (RNRs)
Ruskoski and. Boal. J. Biol. Chem. (2021) 297(4) 101137. DOI:https://doi.org/10.1016/j.jbc.2021.101137. CC-BY license (http://creativecommons.org/licenses/by/4.0/).
Ribonucleotide reductases (RNRs), also called ribonucleoside-diphosphate reductase, catalyze the oxidation of the C2'-OH on the ribose ring to C2'-H through a free radical mechanism for the oxidation of for all NDPs including ADP, GDP, CDP and UDP (which converts to dUDP and in a different reaction to dTDP). The reaction is as follows:
[thioredoxin]-dithiol + ribonucleoside 5'-diphosphate ↔ [thioredoxin]-disulfide + 2'-deoxyribonucleoside 5'-diphosphate + H2O
To refresh your mind, thioredoxin is a small protein (12K) that is part of a complex with thioredoxin reductase and thioredoxin-interacting protein. It has two key sulfhydryls at the active site which act as reducing agents as they get converted to a disulfide as shown in Figure \(\PageIndex{12}\).

Figure \(\PageIndex{12}\): Reduced thioredoxin and its oxidized form
This single enzyme is so critical to cellular life that we will examine it more closely.
There are several classes of these enzymes (Ia-Ie, II, and III). The class I enzymes generally use a di-transition metal complex as a cofactor while class II uses adenosylcobalamin. We will focus on class I enzymes, which have two subunits, called α and β or M1 and M2, respectively. The NDP binds in an α/M1 subunit active site which is developed only in the dimer. The β/M2 subunit is often referred to as the radical-generating subunit as it contains the transition metal complex that generates a free tyrosyl radical cation critical to the reaction.
Almost all class I ribonucleotide reductases (RNRs) use transition metal ions located in the β/M2 subunit in the catalytic cycle for the dehydroxylation of the 2' OH on the ribose ring of the nucleotide. The metal ion complex a β/M2 subunit tyrosine to a tyrosine free radical cation which oxidizes an active site cysteine in the α/M1 subunit to form a thiol radical cation (Cys•+), called a thiyl radical. This abstracts a H• from the 3'C on the ribose of the substrate, forming a 3'C radical cation. This facilitates a dehydration reaction which leads to the dehydroxylation of the 2' OH, regenerating the thiyl radical. Reducing equivalents to restore the catalytic function of the enzyme come from the oxidation of a thioredoxin disulfide bound in the other subunit of the protein or a formate.
Given the importance of these enzymes, they must be highly regulated. There are two regulatory sites:
- a specificity site: determines nucleotide (NDP) specificity
- an activity site: regulates catalytic activity
The specificity and activity sites are in the α/M1 subunit where allosteric regulators dNTPs and ATP bind to different sites. When ATP is bound, the enzyme uses CDP and UDP as substrates. When dGTP is bound, ADP is the preferred substrate. Finally, when dTTP is bound, GDP is the preferred substrate. The enzyme is inhibited by dATP binding to the actual active site.
Figure \(\PageIndex{13}\)s shows an abbreviated mechanism and cartoon showing the activities of the two subunits.

Figure \(\PageIndex{13}\): Abbreviated mechanism and a cartoon showing the activities of the two subunits. in class I RNR. A, universal mechanism for nucleotide reduction in RNRs. B, diagram of the steps involved in radical translocation in class I RNRs. Ruskoski and. Boal. J. Biol. Chem. (2021) 297(4) 101137. DOI:https://doi.org/10.1016/j.jbc.2021.101137. CC-BY license (http://creativecommons.org/licenses/by/4.0/)
Radical formation starts at tyrosine 122 in the metal center site in the β/M2 subunit. Electron transfer then occurs across the two subunits from a very distant active site Cys 439 in the α subunit. which enables the formation of the thiyl radical cation (Cys•+).
The structure of an E. Coli Type IA enzyme with bound ligands has been determined after much effort that involved trapping of a long-life intermediate. It required replacement of Tyr 122 in both β chains with 2,3,5-trifluorotyrosine, which allowed the structure to be determined by cyro-EM. Tyr 122 in the β chain forms starts the process of electron transfer as it becomes the Tyr 122.+ radical cation.
A detailed mechanisms showing both electron transfer to Y122.+ and accompanying proton transfer is shown in Figure \(\PageIndex{14}\).

Figure \(\PageIndex{14}\): Pathway of electron and proton transfers for the formation of the thiyl radical cation (Cys•+). after Kang et al. Science (2020). DOI: 10.1126/science.aba6794)
The path for electron flow from the sulfur of C439 in the β subuit to regenerate Y122 is shown. That electron transfer occurs over a very long distance of 35 Å as it hops from the original sulfur donor to the acceptor.
Figure \(\PageIndex{15}\) shows an interactive iCn3D model of the holocomplex of E. coli class Ia ribonucleotide reductase with GDP and TTP (6W4X).
.png?revision=1&size=bestfit&width=457&height=411)
Figure \(\PageIndex{15}\): Holocomplex of E. coli class Ia ribonucleotide reductase with GDP and TTP (6W4X). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...71ubUDRovXURG8
The structure is a tetramer of two α chains (different shades of gray) and two β chains (different shades of cyan). The GDP and TTP in the α subunit and the Fe cluster in the β subunit are shown in CPK spacefill and labeled. The key side chains in the α and β chains that participate in electron transfer over the 35 Å distance are shown in CPK-colored sticks and labeled. The Fe2+ ions are in the form of a μ-oxo-diron complex (2 Fe ions coordinated by 1 oxide).
Figure \(\PageIndex{16}\) shows specificity and catalytic sites of the biologically active tetramer form for Class 1 RNR as well as the transition metal cofactors (Fe, Mn, or both) in various Class 1 RNRs.

Figure \(\PageIndex{16}\): Quaternary structure of the active holoenzyme complex in class I RNR (PDB accession code 6W4X). Insets show the location of the active site in the catalytic α subunit (middle top) and the metallo- or radical cofactor (middle bottom and far right) in the β subunit
The tyrosyl free radical forms on binding of dioxygen to the transition metal ion center electron and subsequent loss of an electron from tyrosine, as shown in Figure \(\PageIndex{17}\):

Figure \(\PageIndex{17}\): Cofactor assembly mechanisms for class I RNRs. Manganese-dependent enzymes are highlighted in purple. Superoxide-dependent RNRs are highlighted in hot pink. Subclasses that require a NrdI activase are indicated with a yellow box. Metal-centered Cys oxidants shown in green and Tyr-derived radical Cys oxidants shown in blue.
The mechanisms of cofactor actions are not fully understood. O2 initially adds to the Fe2+/Fe2+ cluster which forms a peroxo-Fe3+/Fe3+ intermediate.
Structural features of the active site for different class I RNRs along with redox-active transition metal ions are shown in Figure \(\PageIndex{18}\).

Figure \(\PageIndex{18}\): Comparison of metal-binding sites in class I RNRs
The PDB codes for each structure are as follows: A, (3N3A), B, 4M1I), C, (6CWP), and D (6EBO). Water molecules are shown as red spheres.
Now let's look at the regulation of class IA RNRs in more detail. Let's consider perhaps the most important allosteric regulators which bind 15 Å from the active site which affect RNR enzymatic activity:
- dATP: inhibits RNRs when it binds to the α subunit
- ATP: reverses the inhibition by dATP
dATP/ATP also affects RNR enzyme specificity as they tilt the preference of RNR towards pyrimidine substrates, where TTP and dGTP promote purine substrate binding. These same rules apply to human and E. coli RNRs, with the locations of the active sites also being the same.
The allosteric regulators appear to affect the quaternary structure of the enzyme.
In E. Coli, the binding of dADP converts the structure of the enzyme from an active α2β2 form to an inhibited α4β4 ring structure. When dATP is bound, a "cone" domain in α forms interactions with the β subunit, leading to the formation of a dimer of the α2β2 tetramer. ATP reverses this effect by displacing dATP and pushing the equilibrium towards the active α2β2 form. This is shown in Panel A of Figure \(\PageIndex{19}\).

Figure \(\PageIndex{19}\): Comparison of mechanisms of allosteric regulation of activity for E. coli and human RNRs. Brignole et al. 3.3-Å resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound eLife 7:e31502. https://doi.org/10.7554/eLife.31502. Attribution 4.0 International (CC BY 4.0).
Panel B above illustrates the regulation of the human enzyme, which appears to be quite different. In the absence of either dATP or ATP, RNRs exist just as α2 dimers. On binding either dATP or ATP, the α2 dimer is converted to an α6 (a trimer of dimers) structure. Both are inactive in the absence of β subunits (which provide the metal cofactor site)
- When β2 is added to the dATP bound α6 hexamer, the hexamer becomes stabilized, but the resulting complex is inhibited.
- When β2 is added to the ATP bound α6 hexamer, the hexamer becomes destabilized and into smaller structures which are active.
Hence the ratio of cellular dATP/ATP changes the aggregation state of the RNR and hence its activity in both E. Coli and humans.
Figure \(\PageIndex{20}\) shows an interactive iCn3D model of the Human ribonucleotide reductase large subunit (alpha) with dATP and CDP (6AUI).
_with_dATP_and_CDP_(6AUI).png?revision=1&size=bestfit&width=310&height=321)
Figure \(\PageIndex{20}\): Human ribonucleotide reductase large subunit (alpha) with dATP and CDP (6AUI). (Copyright; author via source). Click the image for a popup or use this external link:https://structure.ncbi.nlm.nih.gov/i...w1sFwwYSAD4nG6
Each of the α subunits in the hexamer is shown in a different color. dATPs (allosteric inhibitors) are shown in spacefill, red. CDPs (substrates) are shown in spacefill yellow.
The hole in the middle of the structure prevents β2 from binding in a catalytically productive fashion, so the structure is inactive.
A specific loop, loop 2, assists in the determination of RNRs specificity. It is between the base of dATP and the base of the substrate CDP. The backbone of the loop interacts with the adenine base and orients Gln 288 in the active site to interact with the cytosine of the substrate CDP. In both systems, backbone atoms of this loop ‘read out’ the adenine base and position Gln288 into the active site to recognize the cytosine of CDP. Figure \(\PageIndex{20}\) gives further details on the origin of substrate specificity.

Figure \(\PageIndex{21}\): Determinants of substrate specificity are conserved from E. coli to humans.
Panel (A) shows residues of human α (blue) interacting with CDP (carbons in orange) in the active site and dATP (carbons in yellow) in the specificity site. Density for CDP in orange mesh and for dATP in the yellow mesh.
Panel (B) zooms in on dATP in the specificity site. Water molecules and oxygen atoms are in red, nitrogen in blue, magnesium in green, and phosphate in gold.
Panel (C) zooms in on CDP in the active site.
Panel (D) shows an overlay of human α from the α6 EM structure in blue with E. coli α from the α4β4-CDP-dATP cocrystal structure in gray (PDB: 5CNS) shows a nearly identical loop 2 conformation positioning Gln288 and Arg293 (Gln294 and Arg298 in E. coli).
Panel (E) shows an overlay of human α from the α6 EM structure in blue with the crystal structure of human α with N- and C-termini truncated (residues 77–742) cocrystallized with dATP in tan (PDB: 2WGH) shows similar positioning of dATP but an altered conformation of loop 2 in the absence of bound CDP. The CDP shown is from the α6 EM structure.
Panel (F) shows an overlay of human α from the α6 EM structure in blue with equivalent residues of yeast α structure with CDP and AMPPNP in brown (PDB: 2CVU) and shows a conformation of loop 2 that is distinct from that seen in structures of E. coli and human α
Transcriptional regulation of RNRs
Given the importance of this key enzyme, it should come as no surprise that its levels are regulated at the transcription level. As the activity of RNR is determined by its polymeric quaternary state, the transcriptional activation of the genes for RNRs is controlled in bacteria by quaternary states of the RNR-specific transcriptional repressor NrdR. The transcription factors bind to a specific DNA sequence called an NrdR box, which precedes the start site of transcription for RNR, where RNA polymerase binds.
The NrdR protein acts to repress the synthesis of RNR genes. Its aggregation state is controlled by dATP/ATP ratios. When dATP is high, the NrdR binds to DNA and represses the synthesis of the RNR gene. In contrast when ATP is high, the protein does not bind to DNA and hence it can not repress transcription. The association /dissociation of the repressor is controlled by the aggregation state of NrdR. When abundant, NrdR exists as a 12-mer complex with two molecules of ATP bound per monomer. This ATP-bound 12-mer can't bind DNA, so transcription of the gene for RNR is not repressed. As dATP increases, one ATP is displaced so each monomer has 1 dATP and 1 ATP bound. This causes the NrdR to covet to an 8-mer. A 4-mer (tetrameric) version of this protein binds to the NrdR box sequence at the start of the RNR gene, repressing its synthesis.
Figure \(\PageIndex{22}\) shows the dodecameric, octameric, and tetrameric structures of NrdR and their functions.

Figure \(\PageIndex{22}\): Mechanism of NrdR function involving dodecameric, octameric, and tetrameric structures. Rozman Grinberg, I., Martínez-Carranza, M., Bimai, O. et al. A nucleotide-sensing oligomerization mechanism that controls NrdR-dependent transcription of ribonucleotide reductases. Nat Commun 13, 2700 (2022). https://doi.org/10.1038/s41467-022-30328-1. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
Panel (a) shows a surface representation of the cryo-EM maps for the dodecameric, octameric, and DNA-bound tetrameric NrdR structures.
Panel (b) shows a cartoon representation of the ATP-loaded NrdR tetramer (left) and the dATP/ATP-loaded tetramer (right). Chains A, B, C, and D are colored in beige, green, pink, and blue, respectively.
Panel (c) shows the interface between the ATP cones in chain A (beige) and chain B (green) in the ATP-loaded dodecamer.
Panel (d) shows the dATP/ATP-loaded tetramer.
Panels c, and d were made from the same perspective, based on an alignment of the ATP-cones in chains A and B in both structures.
Figure \(\PageIndex{23}\) shows an interactive iCn3D model of the Streptomyces coelicolor dATP/ATP-loaded NrdR in complex with its cognate DNA (7P3F).
.png?revision=1&size=bestfit&width=686&height=352)
Figure \(\PageIndex{23}\): Streptomyces coelicolor dATP/ATP-loaded NrdR in complex with its cognate DNA (7P3F). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...2u3HMRPmJHe9X9
The monomers of the NrdR tetramer are shown in different colors. The yellow spheres are dATP and the orange ones are ATP.
Pyrimidine Salvage Pathway
There is also a salvage pathway as shown in Figure \(\PageIndex{24}\).

Figure \(\PageIndex{24}\): Pyrimidine salvage pathway (after Wang et al. Frontiers in Oncology, 11 (2021). https://www.frontiersin.org/article/...nc.2021.684961. DOI=10.3389/fonc.2021.684961
Cells at rest use the salvage pathway reactants derived from nucleic acid degradation generic nucleoside pools.
Nucleotide Degradation
Purines
The pathway for purine degradation is shown in Figure \(\PageIndex{25}\).

Figure \(\PageIndex{25}\): Purine degradation
Pyrimidine Degradation
The pathway for pyrimidine degradation is shown in Figure \(\PageIndex{26}\).

Figure \(\PageIndex{26}\): Pyrimidine degradation