
28.3: The Next step - The Kinome and Activation of Kinases at the Cell Membrane
- Page ID
- 75912


In the last section, we studied how the cell surface GPCRs indirectly activate membrane-bound enzymes adenylyl cyclase and phospholipase C (the GPCRs) and how RTKs become active enzymes (kinases) themselves on ligand-induced dimerization. We will now explore what happens next in signaling by examining the products of these activated enzymes and how they continue the signaling process. The products of adenylyl cyclase (cAMP) and phospholipase C (diacylglycerol - DAG - and IP3) are second messengers. These activate key protein kinases in the cell. The products of the activated RTKs are phosphoproteins (including itself) that are phosphorylated on tyrosines by the RTK. Target proteins bind to the autophosphorylated RTKs through SH2 domains on the target protein. First, we will explore protein kinases and their mechanism in general.
The Kinome
There are 518 different protein kinases (about 1.7% of the human genome) and as a group, they are the key players in most eukaryotic signaling pathways. Collectively they are called the kinome. They help regulate every aspect of cell life including metabolism, cell growth, differentiation, and division, as well as programmed cell death. In humans, most (388) are Ser/Thr kinases. 90 are Tyr kinases and 40 are classified as atypical. In this section, we will focus on the AGC kinases (60 in total) which include Protein Kinase A (PKA), Protein Kinase C (PKC), and also Protein Kinase B (PKB, better known as AKT. We'll wait for another section to explore Protein Kinase G (PKG) which is activated by cGMP, not AMP. We'll also study the receptor Tyr kinases (RTKs)
Luckily kinases can be grouped into families based on structure and mechanism. The ones directly activated by the second messengers cAMP (Protein Kinase A) and DAG (Protein Kinase C) are members of one family of kinases the AGC Protein Kinase family. The clustered families of protein kinases are shown in Figure \(\PageIndex{1}\).
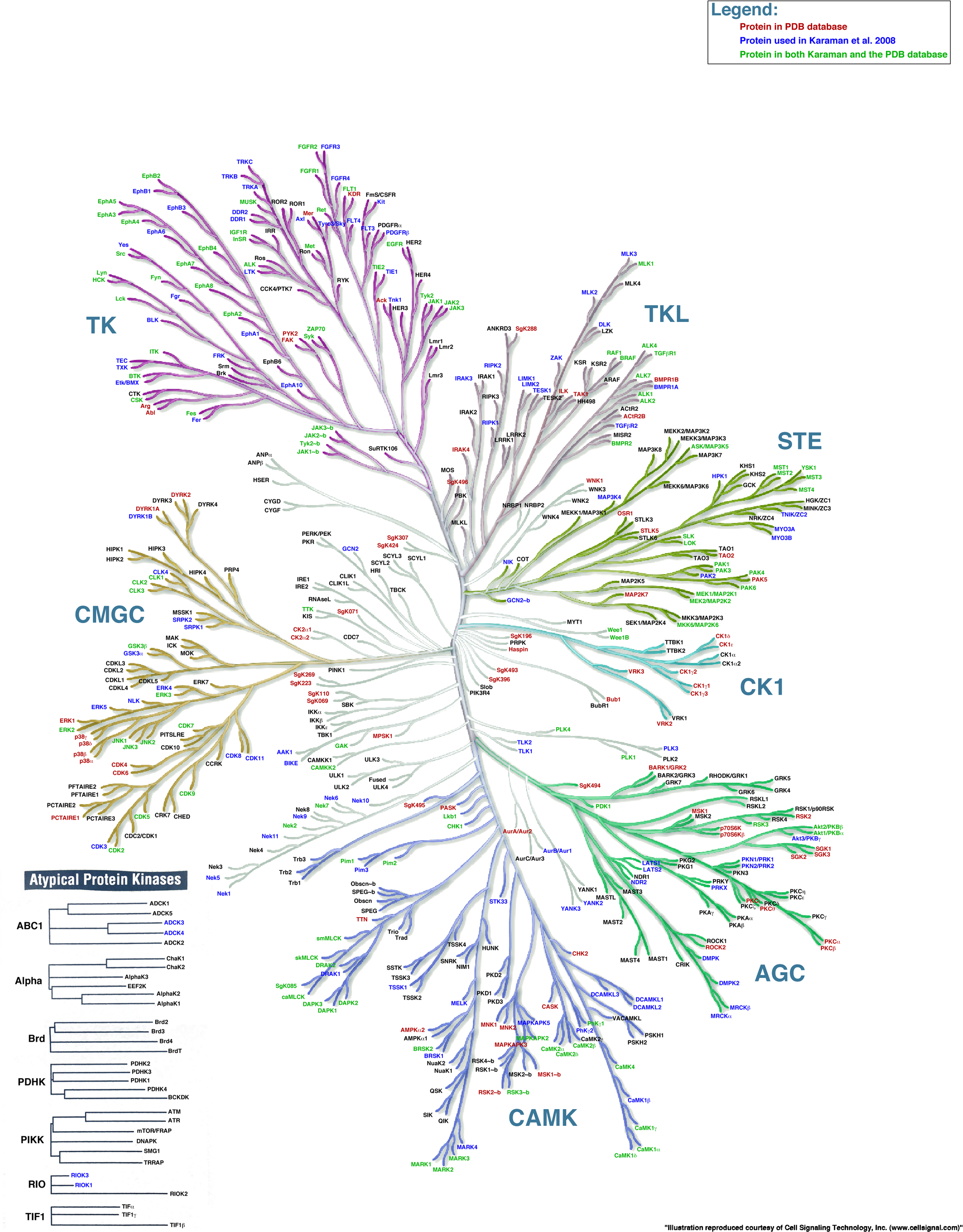
Figure \(\PageIndex{1}\): The family of protein kinases. https://peerj.com/articles/126/. Illustration reproduced courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com)
Luckily, most of the kinases, which catalyze the phosphorylation of the OH-containing side chains (Tyr, Ser, and Thr) of proteins have similar catalytic sites. Given that phosphorylation of proteins often acts as an on/off switch for protein activity and function, it makes great sense that the catalytic site of protein kinases is not available for substrate binding and/or catalysis in the off state. Protein kinases have catalytic and activation loops and conformation changes on activation of protein kinases usually involve movement of the activation loop and rearrangement of the catalytic residues. These conformational changes are often initiated by phosphorylation by another upstream active protein kinase of key groups in the activation loop! Figure \(\PageIndex{2}\) shows another representation of the different families of protein kinase and their common structural features.

Pane (A) shows the nine eukaryotic and one atypical protein kinase groups with their numbers of kinases, structures, and inhibitors (shown in black) as well as mutations (in red) and SNPs (in green). Here, the atypical protein kinases are defined as all non-eukaryotic protein kinases including also the nonprotein kinase-like kinases. The middle of Panel (B) shows the activation and catalytic loops and other common features found in the active sites of protein kinases. The left and right parts of Panel (B) show two different kinases. The protein kinase domain of epidermal growth factor receptor (EGFR) is shown on the left and the Ser/Thr protein kinase domain of mTOR (mammalian or mechanistic Target Of Rapamycin), a protein we will explore in a separate section, is shown on the right.
AGC Kinases
These serine/threonine kinases are activated by the second messengers cAMP (A Kinase), cGMP (G kinase, which we will see later), and by diacylglycerol (DAG) formed by activation of phospholipase C. Figure \(\PageIndex{3}\) shows the generic structure of active kinase domains (including tyrosine kinases)

A generic kinase has an N-terminal and C-terminal lobe between which the substrates ATP and the target protein containing the key serine, threonine, or tyrosine side chain bind. In the active conformation, Glu 91 (E91) in the αC-helix in the N-lobe forms a salt-bridge (ion-ion interaction) with Lys72 to position it so it can stabilize the binding of the ATP through ion-dipole and H-bond interactions. In the active state, Thr 197 (T197) in the activation loop has been phosphorylated (represented by pT197) by an upstream kinase allowing a less flexible conformation of the loop. This facilitates optimal repositioning of active site side chains in the catalytic loop allowing substrate access and catalysis. Asp 166 (D166) in the catalytic loop acts as a general base in abstracting a proton from the target Ser, Thr or Tyr. Arginine 165 (R165) in the catalytic loop forms a salt bridge to phosphotyrosine 197 (pT197), stabilizing the catalysis-competent conformation of the activation loop. Aspartic acid 184 (D184) plays a key role in stabilizing the Mg2+ that itself stabilized the negative charges on the phosphates of ATP and in the developing transition state.
Most kinases are also classified as RD kinases since they have a key arginine (R)- aspartic acid (D) sequence in the catalytic loop. The normal protein substrate for an AGC kinase would be a protein with a Ser or Thr projecting into the active site for phosphorylation by bound ATP. The normal sequence for phosphorylation in protein targets of PKA is Arg-Arg-X-Ser/Thr-X (or more fully as XR(R/K)X(S/T)B where B is a hydrophobic amino acid). Again we will focus on three AGC kinases here, PKA, PKC, and AKT (PKB), and defer our discussion of PKG to another section.
cAMP-dependent Protein Kinase A - PKA
Before we study PKA, the formation of the second messenger cAMP is presented in review in Figure \(\PageIndex{4}\).

PKA contains a kinase domain with short N- and C-terminal extensions, making it a prototypical and simple model for study. In the absence of the second messenger cAMP, this prototypic AGC kinase exists as a holo-heterotetramer (or dimer of a heterodimer). It contains two catalytic kinase subunits (C) and two regulatory subunits (R). cAMP produced upon GPCRs activation of adenylyl cyclase binds to the regulatory subunits, causing them to dissociate from the heterotetramer, freeing the catalytic subunits for activity. This is illustrated in Figure \(\PageIndex{5}\).

The cAMP serves as an allosteric activator of protein kinase A.
Figure \(\PageIndex{6}\) shows the actual structures of the protein kinase A RIIb tetrameric holoenzyme (3TNP)

The inhibited catalytic subunits are shown in red and the two regulatory subunits are shown in cyan.
Figure \(\PageIndex{7}\) shows only one catalytic subunit of protein kinase A as it goes from the more closed inactive holoenzyme (tetramer, 3tnp) containing the regulatory subunits (not shown), to the more open apo-form (monomer, 1J3HA), without the regulatory subunit. The conformational change opens the free catalytic subunit to the substrate (protein and ATP) binding.
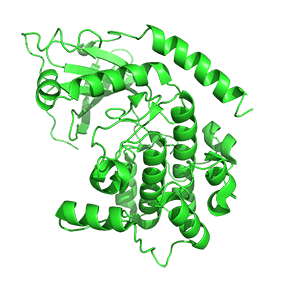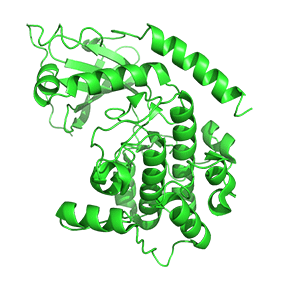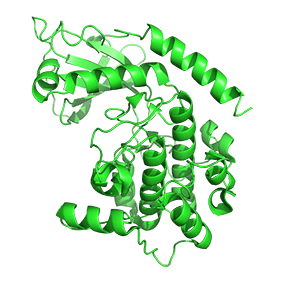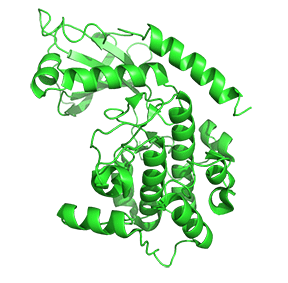
Side chains required for MgATP binding and phosphoryl transfer are pre-formed in the apo form but some changes still occur on the binding of substrate.
Figure \(\PageIndex{8}\) shows an interactive iCn3D model of the mouse catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor (1ATP)
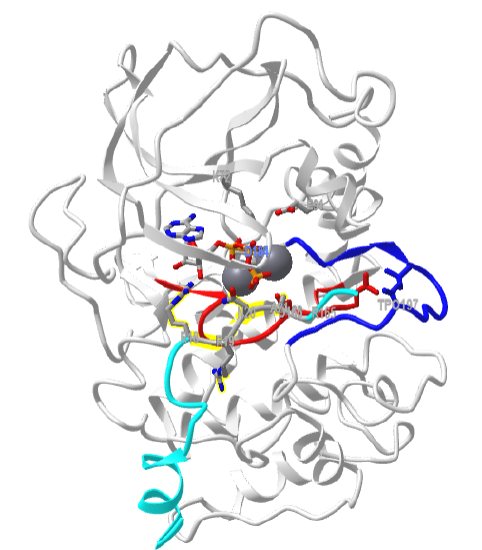
Zoom into the model to see each of the labeled residues. All of the features shown in Figure \(\PageIndex{3}\) are displayed in this model. The activation loop is shown in dark blue and the catalytic loop is shown in red. The peptide inhibitor is shown in cyan. The normal target motif for phosphorylation by Protein Kinase A (Arg-Arg-X-Ser/Thr-X) has been replaced in this model by Arg-Arg-X-Ala-X. Why does this make the peptide an inhibitor instead of a substrate?
Many primary signals activate adenylate cyclase through GPCR and produce cAMP as a second messenger. These include corticotrophin, dopamine, epinephrine (β-adrenergic), follicle-stimulating hormone, glucagon, many odorants, prostaglandins E1and E2, and many tastants. All of these would activate protein kinase A. Some enzymes regulated by cAMP-dependent phosphorylation by PKA are shown in Table \(\PageIndex{1}\) below
| Enzyme | Pathway |
| Glycogen Synthase | Glycogen synthesis |
| Phosphorylase Kinase | Glycogen breakdown |
| Pyruvate Kinase | Glycolysis |
| Pyruvate Dehydrogenase | Pyruvate to acetyl-CoA |
| Hormone-sensitive Lipase | Triacylglycerol breakdown |
| Tyrosine Hydroxylase | Synthesis of DOPA, dopamine, norepinephrine |
| Histone H1 | Nucleosome formation with DNA |
| Histone H2B | Nucleosome formation with DNA |
| Protein phosphatase 1 Inhibitor 1 | Regulation of protein dephosphorylation |
| CREB | cAMP regulation of gene expression |
| PKA consensus sequence | XR(R/K)X(S/T)B (B = hydrophobic amino acid) |
Table \(\PageIndex{1}\): Proteins phosphorylated by Protein Kinase A.
An example of how epinephrine (a flight/fight hormone) can lead to the breakdown of glycogen (your main carbohydrate reserves in muscle and liver) is shown in Figure \(\PageIndex{9}\).

A cascade of events starts with the binding of the hormone to its receptor, followed by the activation of adenylate cyclase and the formation of the second messenger cAMP. This activates PKA, which phosphorylates and activates the enzyme phosphorylase kinase. Following the naming convention we discussed earlier, phosphorylase kinase is a protein kinase whose target enzyme is another enzyme called glycogen phosphorylase, which is NOT a kinase. When active, glycogen phosphorylase uses inorganic phosphate (Pi) as a nucleophile in a phosphorolysis reaction to cleave glucose from the end of glycogen polymers forming glucose-1-phosphate. This is the first step in the mobilization of glycogen as an energy reserve. No wonder it is so tightly regulated by this complicated signaling pathway.
The second messengers DAG and IP3 and their activation of Protein Kinase C
The activation of protein kinase C (PKC, a Ser/Thr kinase member of the ACG protein kinase family) is very similar to that of protein kinase A. To start, an extracellular signal molecule binds to a GPCR receptor (again with no intrinsic enzyme activity), causing a conformational change in the receptor that propagates through the membrane to its intracellular domain. That then activates the exchange of GTP for GDP in the alpha subunit of the bound heterotrimeric G protein, which contains the special Gαq subunit. The Gαq subunit dissociates and binds to the membrane protein enzyme phospholipase C (not adenylyl cyclase). Once activated, it cleaves the phospho-head group from the membrane phosphatidyl inositol - 4,5-bisphosphate (PIP2) into two, second messengers - diacylglycerol and inositol trisphosphate (IP3). These products are shown in Figure \(\PageIndex{10}\).

The formation of the second messengers IP3 and DAG is presented n Figure \(\PageIndex{11}\). Note that phospholipase C is a peripheral membrane protein bound to the inner leaflet of the cell membrane.

Diacylglycerol binds to and activates protein kinase C (PKC). The IP3 binds to ligand-gated receptor/Ca2+ channels on internal membranes, leading to an influx of calcium ions into the cytoplasm. The released calcium ions also activate PKC. These steps are illustrated in Figure \(\PageIndex{12}\) in more detail below. The activation of the peripheral membrane phospholipase C (PLC) is very similar to that of the integral membrane protein adenylyl cyclase, both of which produce second messengers.

Figure \(\PageIndex{12}\): GPCR activation of phospholipase C, generation of second messengers IP3, DAG, and Ca2+ ions, and downstream activation of Protein Kinase C
You might also expect the regulation of the activation of protein kinase C (PKC) activity to be similar to the regulation of the activation of protein kinase A (PKA). It is, but with a major difference. In contrast to the structure of PKA, which cycles between an inactive R2C2 (R is the regulatory and C the catalytic subunit) and an active separated form, PKC is a single chain that has a regulatory, domain, and catalytic domain as shown in Figure \(\PageIndex{13}\). Variants of PKC are also shown.

Protein kinase C was named "C" because of its activation by Ca2+ ions, but you could also remember it because it requires the upstream activation of phospholipase C.
There are more than 500 PKCs divided into 15 subgroups and their downstream functions lead to gene expression. They all have 4 common domains, C1-C4. C1 and C2 are the functional regulatory domain and C3 and C4 are in the overall catalytic domain. They have the following activities.
- C1 binds diacylglycerol (DAG) and phorbol esters (commonly known activators or PKC)
- C2 binds Ca2+, another second messenger, which activates the protein; Novel PKCs use DAG and phosphatidyl ethanolamine but not Ca2+ to activate the protein, while atypical ones use only DAG.
- C3 binds ATP
- C4 binds target proteins for phosphorylation.
The structure of the phorbol 12-myristate 13-acetate, which activates PKC, is shown in Figure \(\PageIndex{14}\).

In a novel way to keep the protein inactive, it contains a small peptide sequence (a pseudosubstrate) that mimics the amino acids around the target phosphorylation site (with the target Ser/Thr). This binds in the active site in the inactive form of PKC like a competitive inhibitor and prevents PKC activity.
PKCα is found in the cytoplasm, cell membrane, nucleus, and mitochondria. Its activation by DAG suggests it becomes localized to membrane surfaces. The protein is conformationally very flexible (note the hinge domain in Figure \(\PageIndex{13}\)) so it has been difficult to get detailed structures.
Figure \(\PageIndex{15}\) shows an interactive iCn3D model of the PKC (alpha)-C2 domain complexed with Ca2+ and PtdIns(4,5)P2 (IP3) (PDBID 3GPE). The hydrogen bonds and ion-ion interaction between the C2 domain and IP3 are detailed and labeled.
.png?revision=1&size=bestfit&height=435)
Protein kinase C is targeted to the membrane (as shown in Figure \(\PageIndex{12}\)), but it lacks the Pleckstrin Homology (PH) which most phospholipase Cs use to interact with PIP2 in the membrane.
Before activation of PKC by DAG and Ca2+, it must be phosphorylated sequentially. Before these phosphorylations, PKC loosely binds to the membrane with the activation loop open to phosphorylation by kinases. These are the subsequent steps:
- phosphorylation of Thr500 (PKC βII)) in the activation loop of PKC by an upstream kinase PDK1 (a kinase which also phosphorylates other AGC kinases such as AKT discussed below). This kinase can bind to the exposed activation loop
- autophosphorylation at the C-terminus of PKC, which leads to conformational positioning of side chains needed for catalysis and substrate binding, and access to the substrate binding site
PKC engages in a very dynamic cycle. It starts as the inactive cytoplasmic form that is autoinhibited by its pseudosubstrate sequence. It then moves to the membrane where the autoinhibition is relieved. All of this requires flexibility which makes it difficult to determine its structure. Figure \(\PageIndex{16}\) shows how the inactive cytoplasmic form of PKC becomes activated at the cell membrane.

The regulatory domain, which contains the C1 (DAG binding) and C2 (Ca2+) domains binds to the membrane freeing and activating the kinase domain on the release of the bound internal pseudosubstrate.
Figure \(\PageIndex{17}\) the optimal consensus sequence flanking both sides of the phosphorylation site in target proteins (based on model peptides) and the sequence of the internal pseudosubstrate motif for several PKCs.
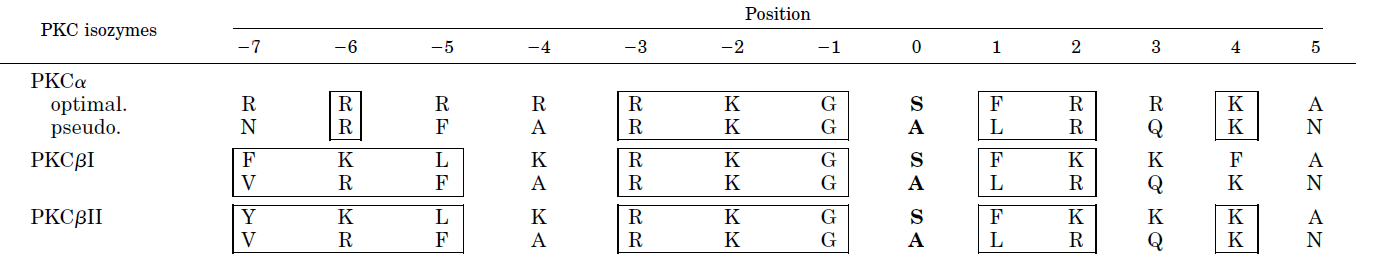
Boxed amino acids show structural similarity between the target and internal pseudosubstrate. Position 0 indicates the serine that is phosphorylated in the target. Note that it is replaced with alanine in the pseudosubstrate. Also, note the abundance of positively charged side chains.
Figure \(\PageIndex{16}\) also shows that phosphorylation of key side chains facilitates the activation of the enzyme. The upstream kinase 3-phosphoinositide-dependent protein kinase 1 (PDPK1 or PDK1) phosphorylates the key Ser/Thr in the activation loop as we discussed above. PDK1 is a "master" Ser/Thr kinase which phosphorylates and activates many proteins, including PKA, PKC, and protein kinase B (which we will explore below).
Figure \(\PageIndex{18}\) shows an interactive iCn3D model of the Protein Kinase C beta II (3PFQ)

The C2 domain (magenta)has two bound Ca2+ ions (gray spheres) and interacts with the bottom leaflet of the cell membrane. The C1 domain (purple) has two bound Zn2+ ions. The N lobe of the kinase domain is shown in green and the C lobe is shown in brown. A nonhydrolyzable ATP analog, AMPPNP (ANP), is shown in spacefill between the N and C lobes. There is an additional NFD helix preceding the C-terminal tail which can adopt two positions, one which confers low activity and the other high PKC activity. The Phe629 in this region is out of the active site in the low activity form and in it and interacting with the adenine of ATP in the high activity form.
Some signals that activate phospholipase C and make IP3 and diacylglycerol include acetylcholine (a different class than the type located at the neuromuscular junction that we discussed in the last chapter section), angiotensin II, glutamate, histamine, oxytocin, platelet-derived growth factor, vasopressin, gonadotropin-releasing hormone, and thyrotropin-releasing hormone.
In the inactive form of PKC, the arginine-rich basic autoinhibitory pseudosubstrate interacts with acidic side chains in the substrate binding site.An acidic patch in the substrate-binding site (Figure 6.2). When PKC is activated by phosphorylation of the regulatory domain, it phosphorylates arginine-rich sites in protein substrates. This also releases pseudosubstrates from some target inactive protein kinases, which allows them to become active kinases in turn. PKC physiological substrates include receptors, cytoskeleton proteins, protein kinases, proteases, and nuclear proteins
The Ca2+ ions also act as second messengers. The calcium ions bind to the calcium-modulatory protein, calmodulin, which binds to and activates the calmodulin-dependent kinase (CAM-PK), which we will discuss later. Some kinases regulated by calcium and calmodulin include myosin light chain kinase, PI-3 kinase, and CAM-dependent kinases. Ca/CAM also regulates other proteins which include: adenylate cyclase (brain), Ca-dependent Na channel, cAMP phosphodiesterase, calcineurin (phosphoprotein phosphatase 2B), cAMP gated olfactory channels, NO synthase, and plasma membrane Ca/ATPase.
Protein Phosphorylation by activated Receptor Tyrosine Kinases (RTKs)
Figure \(\PageIndex{19}\)s shows the dimeric structure of RTKs driven by extracellular signal binding.

Table \(\PageIndex{2}\) below shows the classification of RTKs into classes and families.

Table \(\PageIndex{2}\):EGFR: epidermal growth factor receptor; InsR: insulin receptor; PDGFR: platelet-derived growth factor receptor; VEGFR: vascular endothelial growth factor receptor; FGFR:fibroblast growth factor receptor; CCK: colon carcinoma kinase; NGFR, nerve growth factor receptor; HGFR: hepatocyte growth factor receptor; EphR: ephrin receptor; Axl: from the Greek word anex-elekto, or uncontrolled, a Tyro3 protein tyrosine kinase; TIE:tyrosine kinase receptor in endothelial cells; RYK: receptor related to tyrosine kinases; DDR: discoidin domain receptor; Ret: rearranged during transfection; ROS: RPTK, expressed in some epithelial cell types; LTK: leukocyte tyrosine kinase; ROR: receptor orphan; MuSK: muscle-specific kinase; LMR: Lemur. Sareshma Sudhesh Dev et al. Front. Pharmacol., 15 November 2021 | https://doi.org/10.3389/fphar.2021.772510. Creative Commons Attribution License (CC BY).
We introduced the activation of RTKs in the previous section and indicated that ligand-induced dimerization led to their activation. Let's expand on that now. As in the case of PKC, the intracellular kinase domains of RTKs are inhibited by specific structures in their chains (cis-autoinhibited). These include the activation loop but in addition C-terminal sequences and the sequence region linking the C-terminal domain with the transmembrane domain. This is called the juxtamembrane region. All of these must be phosphorylated for the activation of kinase activity. The autoinhibition is released on ligand binding and dimerization. The kinase domains can also be allosterically activated by the kinase domain of the dimer. Each domain phosphorylates the cytoplasmic domain of the other, so it's called trans-phosphorylation (i.e. a kinase domain on one monomer does not autophosphorylate itself, which would be called cis-phosphorylation). The now active kinase domains recruit target proteins containing SH2 domains (which bind p-Tyr peptides/proteins) in a fashion that propagates signaling. These include proteins involved in other signaling pathways including MAPK and phosphoinositide-3-kinase (PI3K)/Akt pathways which we will discuss later. A particular phospholipase Cγ (PLC-γ) is also activated by an RTK.
Cancers can arise if the RTK signaling pathways, which control cell growth and division, are overactive. Figure \(\PageIndex{20}\) shows four different mechanisms for the expression and/or activation of RTKs that could lead to cancer.

We will focus on the epidermal growth factor receptor (EGFR) for most of the remaining discussion. One of the most interesting questions is how ligand binding in the extracellular domain of the biotopic integral membrane proteins leads to an intracellular signal in the cytoplasmic domain. It's difficult to imagine such an activation propagating through a single transmembrane helix. We'll discuss how ligand-promoted dimerization of the receptor appears to be the main mechanism of activation of RTKs.
Figure \(\PageIndex{21}\) shows the domain structure of three different RTKs, including the insulin receptor (IR) family. The insulin receptor is also synthesized as a single chain but undergoes proteolysis and interchain disulfide bond formation to give the "dimeric" structure shown below.

The EGFR is a member of the ErbB family which consists of 4 members: EGFR (also called Erb1 or HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). The name ErbB arises from the avian ERythroBlastosis oncogene B). They are also called HER after the Human Epidermal growth factor Receptor). The HER2 gene is often dysregulated in breast cancer. Members of the ErbB family have unique numbers and positions of tyrosine in the C-terminal kinase domains. EGFR has 20, of which 12 are phosphorylated. The EGFR is also a bit unique in that it has only one tyrosine in the activation loop that is phosphorylated but the tyrosine itself is not required for kinase activity. Although we suggested earlier that RTKs are activated on dimerization, studies show that RTKs However, an increasing number of studies demonstrate that RTKs exist as inactive dimers in the absence of the ligand.
As shown in Figure \(\PageIndex{21}\), ErbB receptors have 4 extracellular domains, a transmembrane domain, the juxtamembrane region (about 40 residues), the cytoplasmic kinase domain and a C-terminal extension that gets autophosphorylated and which binds downstream target protein through their SH2 domains. Extracellular domains I ( L1) and III/L2 have β-helix solenoid secondary motifs that bind the ligand. Domains II/CR1 and IV/CR2 are cysteine-rich disulfide bonds. Some fraction (>80% through cross-linking studies) of the ErbB receptors exist as dimers at the cell surface in the absence of ligand, a finding in contrast to the older view that ligand binding is required for dimerization
The structure of the extracellular domains of the free ErbB and ligand-bound EGFR receptors show conformational changes that are required for dimer formation. In the absence of the ligand, a "tether" arm, denoted by an open triangle in domain IV in Figure \(\PageIndex{22}\), is close to a buried "dimerization" arm (asterisk *) in domain II of the extracellular regions of EGFR, ErbB3, and ErbB4, effectively inhibiting dimerization. When the ligand is bound, domains I and III interact, freeing the dimerization domains to interact (** in EGFR). These features are illustrated in Figure \(\PageIndex{22}\).

Figure \(\PageIndex{12}\): Schematic representations of the structures of the extracellular regions of the ErbB family. EGFR, ErbB3, and ErbB4 adopt the tethered conformation in the absence of ligand, while ErbB2 adopts an extended, or untethered, conformation that resembles the ligand-activated, dimerization-competent EGFR protomer in the ligand-bound form of the EGFR dimer, shown at the right. The ‘dimerization arm’ and ‘tethering arm’ are shown by an asterisk * and an open triangle, respectively. Ligands are shown in red. Domains I–IV correspond to the domains shown in Figure \(\PageIndex{21}\). Not drawn to scale. Ichiro N. MaruyamaCells. Cells. 2014 Jun; 3(2): 304–330. doi: 10.3390/cells3020304' (http://creativecommons.org/licenses/by/3.0/). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4092861/
In addition, the tether arms on both monomers now interact in the dimer. Note that the ligand binding region is not involved in dimer formation and is not in the dimer interface, as it is with other RTKs where the ligand is involved in direct contact in the dimer interface. Mutations in the II/IV domains that inhibit their interactions do not lead to receptor activation so ligand binding is still required. ErbB2 exists in an extended conformation (no ligand is known for it) so it is free to interact with another ErbB chain in a heterodimer.
Structural studies now suggest that EGFR kinase dimer has a symmetrical inactive conformation in which the activation loop is packed and occluding the active site. In addition, it has an asymmetrical active one in which the activation loop is unpacked and the active site is open. How is this transmitted across the subunits? It appears that the C-lobe of the "activator/donor" kinase interacts with the N-lobe of the adjacent "receiver/acceptor" kinase which activates it through a conformational change. When the ligand binds, the inactive dimer dissociates, and the asymmetric active dimer results.
Two models have been proposed for ligand-gated activation of the Erb dimers: the dimerization and rotation/twist models, as shown in Figure \(\PageIndex{23}\).

- A. Dimerization model: Ligand binding to the I and III extracellular domains of a monomer lead to dimerization by causing the release of the tether arm between I and III, allowing the dimerization arms to become free and interact with each other. This causes the kinases domains to adopt the active state
- B. Rotation/Twist model: The receptor is already a dimer in the unliganded state with the extracellular region untethered and the intracellular kinase domains in an inactive symmetric state. Ligand binding causes the two dimerization arms to extend, causing a twist in the transmembrane segment. This causes the kinase domains to adopt the active asymmetric state with the activator kinase domain of one monomer activating the receiver kinase domain of the second monomer in the dimer, with each forming an extended conformation.
Figure \(\PageIndex{24}\) shows an interactive iCn3D model of the intracellular dimeric EGFR kinase domains in complex with an ATP analog-peptide conjugate (2GS6)
.png?revision=1)
The two EGFR kinase domains are shown in cyan and magenta. The ATP analogs in each domain (spacefill) are thiophosphoric acid O-((adenosyl-phospho)phospho_-S-acetamidyldiester. The peptide substrates (green stick) are 13-mers with a tyrosine (sticks, labeled Y, minus the OHs) connected to the ATP analog.
Figure \(\PageIndex{25}\) shows an interactive iCn3D model of a single EGFR kinase domain showing N and C terminal lobes in complex with an ATP analog-peptide conjugate (2GS6)
.png?revision=1)
The ATP part of the ATP-peptide conjugate is sandwiched between N-lobe (green) and the C-lobe (brown) just as in PKA. The ATP in the peptide conjugate is shown in spacefill while the peptide is shown in gray. The tyrosine (Y) linked to the peptide is labeled.
Figure \(\PageIndex{26}\) shows an interactive iCn3D model of showing the similarity in structure between the PKA kinase domain (1J3H A chain) and the EGFR kinase domain (2GS6)
_and_the_EGFR_kinase_domain_(2GS6).png?revision=1&size=bestfit&width=440&height=299)
The gray structure by itself is the second kinase domain of the EGFR dimer. The superimposed chains are on the other side. Red indicated identical residue and blue nonconserved in the structural alignment. Zoom in on the aligned sequences in blue and red to show how similar the kinase domains are.
In the next chapter section, we will explore the next downstream effects in signaling, mediated by the second messengers cAMP, DAG, and IP3 and the substrates phosphorylated by the ligand-active receptor tyrosine kinases.
The HER2 receptor can form homo or heterodimers with single chain ErbB1-ErbB4 (same as HER1-HER4). HER receptors exist as both monomers and dimers, either homo- or heterodimers. Rapid dimerization of HER1, HER3, or HER4 occurs if they form heterodimers with HER2. In addition, any ErbB dimer with HER2 leads to strong intracellular signaling compared to other HER heterodimers. Ligand binding to HERI, HER3, or HER4 induces rapid receptor dimerization, with a marked preference for HER2 as a dimer partner. Since noncancer cells have very little HER2, correspondingly few heterodimers of HER2 around found. If HER2 is overexpressed as in HER2+ breast cancer cells, more heterodimers are found, and anomalously-high levels of signaling occur. This resulted in a poorer prognosis for HER2+ breast cancers in the past.
A revolution in breast cancer therapy has changed that situation. Humanized antibodies (human antibodies made in mice cells) to HER2, called trastuzumab (Herceptin) are now used in therapy. Since the antibodies are derived from human genes, they are not targeted as foreign by the immune system. In early-stage HER2+ breast cancers, the antibody trastuzumab (Herceptin) is administered intravenously periodically for one year. The antibody selectively binds to HER2 on breast cancer cells. Once bound, the Fc portion of the bound antibody signals the immune system, leading to the recruitment of immune cells and modulators to the tumor cell, leading to its destruction.
If the cancers are in a later stage or if tumor cells are found in lymph nodes, a variant of the anti-HER2 antibody can be given in which a chemotherapeutic drug is covalently attached to the antibody. The structure of the drug Ado-trastuzumab emtansine (T-DM1), trade name Kaycyla, is shown in Figure \(\PageIndex{27}\).

When the antibody-drug complex binds to the receptor on cancer cells, it is taken up into the cell. The antibody is degraded and the toxic drug is released into the cell sparing all other types of cells (only ones with the receptor on it are targeted). The released drug binds to microtubules comprising part of the internal cytoskeleton of the cell and prevents changes necessary for cell division. HER2 has a very low level of expression in noncancer cells so side effects can occur. However, these are much less server than traditional chemotherapy, which targets all dividing cells. The antibody acts as a homing device bringing the attached chemotherapy predominantly to tumor cells. It can be likened to a smart bomb or a cruise missile guided to just one target.
A better image of DMI and the linker connecting to the antibody is shown in Figure \(\PageIndex{28}\). The toxic chemotherapy drug is chemically linked to the antibody through a non-reducible thioether linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC).
.svg?revision=1)
Figure \(\PageIndex{29}\) shows a mechanism for the actual cross-linking reaction. The reaction of the maytansine derivative through its free thiol and the human antibody (trastuzumab) through a free amine should be readily understandable base on the reactions present in Chapter 2.

The final linker after the departure of the N-hydroxysuccinimide is designated MCC.
The C-MET receptor, illustrated as an example in Figure 19, is an RTK involved in cell proliferation and survival, and as such, alterations in its expression can lead to cancer. Its mature form results from selective proteolysis by furin. Its physiological ligand is hepatocyte growth factor (HGF), a multisubunit protein in its mature form. It also binds a naturally occurring smaller splicing variant of HGF called NK1. The structure of C-MET bound to both HGF and NK1 has been solved. Binding of either lead to dimerization of the C-MET receptor, as illustrated in the cartoon representation of Figure \(\PageIndex{30}\).

The glycosaminoglycan heparin enhances c-MET activation by HGF. This is illustrated in Figure 30 above. Heparin binds between the N domain of HGF I and the IPT1 domain of c-MET II, facilitating the interactions between the two domains. In addition, a long enough heparin chain could bridge both HGF I and II, further strengthening the full complex. Structural representations of the c-MET:HGF asymmetric dimer are shown in cartoon form in Figure \(\PageIndex{31}\).

Panel (a) shows the domain structures of human c-MET and HGF. The proteolytic cleavage site of c-MET is located between Arg307 and Ser308. The proteolytic cleavage site of HGF is located between Arg494 and Val495. C-MET927-LZ and full-length HGF were used for structural determination in this study. The dash boxes indicate the domains that were unsolved in cryo-EM maps. Panel (b) shows the 3D reconstruction of the 2:2 c-MET/HGF holo-complex and the corresponding ribbon representation of this complex fitted into the cryo-EM map at 4.8 Å resolution, shown in two orthogonal views. Panel (c) shows the ribbon representation of the c-MET/HGF holo-complex shown in two orthogonal views
Figure \(\PageIndex{32}\): Shows the structure of the c-MET/NK1 symmetrical dimer.

Nuclear RTKs
What makes signal transduction so complicated yet interesting is the unexpected. It turns out that some RTKs (EGFR, VEGFR, FGFR, IR, and NGFR have been found in the nucleus. It's experimentally easy to localize proteins in cells using immunofluorescence microscopy. It's hard to determine their functions. ErbB-2 is one that is also found in the nucleus. Kinase inhibitors blocked the expression of ErbB-2 in the nucleus suggesting that its kinase activity is required for it to translocate to the nucleus. The structure of the membrane forms of ErbB-2, EGFR, and ErbB-3 appear to be the same as the structure of the nuclear forms.
The carboxy-terminal ends of EGFR and ErbB-4 can activate gene transcription as measured by the expression of luciferase (a fluorescent protein) reporter proteins. Hence they appear to act as transcription factors that bind DNA to promote gene expression. For example, nuclear EGFR increases the expression of cyclin D1 which drives progression through the cell cycle. ErbB-2 appears to activate transcription from the promoter of the gene for cyclooxygenase 2 (COX-2). The protein COX-2 leads to the synthesis of inflammatory prostaglandins. It also increases blood vessel growth and is dysregulated in tumors.
Since proteomic analysis of these growth factor receptors shows no DNA binding motifs or domains, they must promote gene transcription through binding to other protein transcription factors in the nucleus.
Back to AGC Kinase - AKT (Protein Kinase B)
We've just explored the:
- activation of Protein Kinase A (an AGC Kinase) through GPCR signaling, activation of the integral membrane protein adenylyl cyclase, production of the second messenger cAMP, which binds to inactive PKA and leads to its activation
- activation of Protein Kinase C, (an AGC Kinase) again through GPCR signaling, which leads to activation of the peripheral membrane protein phospholipase C, production of the second messengers DAG and IP3 from PIP2, and activation of PKC at the membrane.
- activation of receptor tyrosine kinases (RTKs) leading to autophosphorylation of the cytoplasmic kinase domain, followed by recruitment of downstream signaling proteins through binding the phosphorylated RTKs through the downstream protein's SH2 domain
Now let's explore another AGC kinase called AKT or protein kinase B (PKB) that links signaling through RTKs to phosphoinositol-related signaling in a fashion similar to the link between phospholipase C and Protein Kinase C activities. In the process, we will introduce in this section our first nonprotein kinase involved in signaling. It's a lipid kinase called phosphoinositide 3-kinase (P13K) and it's very important.
Since Protein Kinase B is usually referred to as AKT, we will stick with that abbreviation. As with other AGC kinases, AKT is a Ser/Thr protein kinase. There are three variants, AKT1, AKT2, and AKT3. These are involved in metabolism, growth, and proliferation so they are key players in signaling. It is a key player in the uptake of glucose into cells as it regulates the translocation of the glucose transporter SLC2A4/GLUT4 to the cell surface in response to insulin.
You would expect aberrant expression of these would lead to cancer. The abbreviation AKT appears to derive from "a serine/threonine protein kinase encoded by the oncogene in the transforming retrovirus isolated from the thymoma cell line AKT-8, which is derived from the Stock A Strain k AKR".
As with Protein Kinase C as phospholipase C, AKT is recruited to the inner leaflet of membranes. Recruitment is mediated by its binding through its pleckstrin homology (PH) domain to phosphatidylinositol (3,4,5)-trisphosphate, a modified form of PIP2, abbreviated either as PtdIns(3,4,5)P3 or more simply as PIP3. Note that PIP2 has 3 phosphate groups while PIP3 has 4.) The mechanism of membrane recruitment is similar to that of phospholipase C, which also binds membrane PIP2 through its pleckstrin homology (PH). (This is in contrast to PKC which is recruited through its C1 and C2 domains.)
PIP3 is generated in the membrane from PIP2 by the enzyme phosphoinositide 3-kinase (P13K), a lipid kinase, whose own activation occurs through stimulation of insulin and growth factors receptor tyrosine kinases (RTKs). Class 1 PI3K has a regulatory/adapter subunit (p85) and a 110 kDa catalytic subunit (p110). The regulatory subunit has SH2 domains which recruit it to autophosphorylated RTKs.
Figure \(\PageIndex{33}\) shows how membrane PIP2 can be converted to the second messengers DAG and IP3 by phospholipase C, or to PIP3 by the enzyme phosphoinositide 3-kinase (P13K), which is a lipid kinase.

The binding of AKT (PKB) to inner leaflet PIP3 through its PH domain causes a conformational change that activates AKT for phosphorylation by phosphoinositide-dependent kinase 1 (PDK1) a membrane protein kinase. Once activated. AKT dissociates from the membrane and acts enzymatically in the cytosol and nucleus. The overall activation of AKT is shown in Figure \(\PageIndex{34}\).

.
The blunt arrows in the figure above show inhibition by the enzymes indicated. These (PTEN, PP2A, and PHLPP 1/2) are phosphatases.
- PTEN is lipid phosphatase (phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase), which removes a phosphate from the lipid PIP3.
- PP2A and PHLPP 1/2 are protein phosphatase that removes phosphates added by the kinase PDK1 in the kinase domain and mTORC2 in the C-terminal domain.
Figure \(\PageIndex{35}\) shows an interactive iCn3D model of AKT bound to a novel allosteric inhibitor is shown below (3o96).
2.png?revision=1)
The PH domain (at N-terminus, bind to PIP3) is shown in magenta, the N-lobe in green, and the C-lobe in brown. The activation loop is in dark blue and the catalytic loop is in red. The blue activation loop is shown in two parts as the interior part, which contains T305 (equivalent to T197 in AGC kinases without a PH domain) is too disordered to resolve. Table \(\PageIndex{1}\) shows the numbering of key amino acids and features of generic AGC kinase and the corresponding numbers in AKT. They are different since AKT has an N-terminal PH domain.
| SITE | Generic | Akt (+108) |
|---|---|---|
| N lobe | K72 | K180 |
| N lobe | E91 | E199 |
| C lobe, cat loop | R165 | R273 |
| C lobe, cat loop | D166 | D274 |
| C lobe, act loop | D184 | D292 |
| C lobe, act loop | T197 |
T308* |
| Approx Cat Loop | 163-179 | 271V—287H |
|
Approx Act loop Start DFG (292-294) to APE |
184-200 |
292-308 308Tmiss toAPE end 319 |
The allosteric inhibitor shown in the structure above is especially interesting in that it requires both the PH domain as well as the kinase domains for its effect.
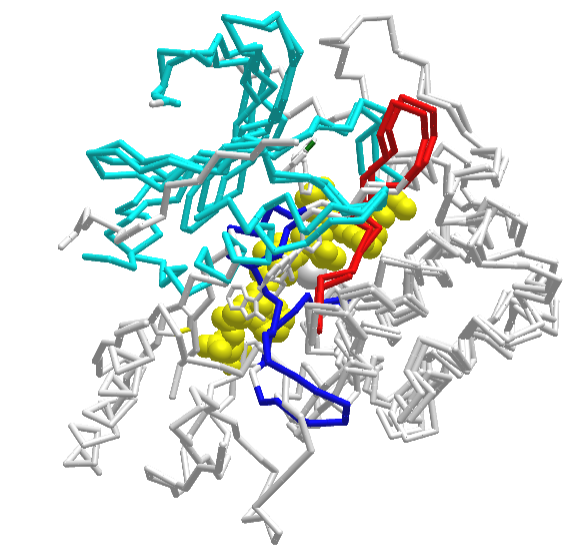
Figure \(\PageIndex{36}\) shows an interactive iCn3D model of the structural overlap between the inactive form (shown above, 3o96 containing a bound allosteric inhibitor) with an active form of AKT(3cqw), which has a bound substrate (from glycogen synthase kinase-3 beta, yellow spacefill).
.png?revision=1&size=bestfit&width=340)
The bound decapeptide substrate (GRPRTTSFAE) in the active form becomes phosphorylated on the Ser chain by active AKT. The N-lobe is shown in cyan, while the catalytic loop is in red and the activation loop is in blue. By pressing the "a" key you can toggle between the inactive 3o96 form and the active 3cqw forms. Note the large change in the blue activation loop.
Figure \(\PageIndex{37}\): below shows a series of coupled equilibria reactions that regulate the activity of AKT1.

The top part of the figure shows the cytoplasmic, nonmembrane-bound form of the enzyme. The PH domain is shown in orange. The top-right figure shows the inactive N- and C-lobes of the kinase loosely interacting with an "out" (or away) conformation of the PH domain with respect to the kinase domains. In the presence of the allosteric inhibitor (green hexagon, green bound ligand), the kinase domains tightly interact with the PH domain in the "in" conformation.
The bottom three structures show AKT bound to the membrane through the interaction of the PH domain with PIP3 (purple). The bottom right kinase domains are identical in representation to the two in the top part of the figure, showing that they are inactive. Only when bound to the membrane is AKT phosphorylated on Thr 308 (red in the bottom middle figure), which activates the enzyme. The bottom left and middle structures show the yellow kinase domains in a different conformation (3cqw), both of which are phosphorylated (red). The active form can bind ATP and protein substrates for phosphorylation. It can also bind ATP analogs which would competitively inhibit the active form of the enzyme by occupying the ATP binding site.
The activation loop in the inhibited form is missing part of its sequence which reflects its disorder. In this state, the loops partially occludes substrate interactions. On phosphorylation of Ser 308 in the activation loop, the loop adopts a different conformation which allows less restricted access to the active. The loop itself on phosphorylation has more local ordering as it shifts away from the active site as seen in the iCn3D model above. Another change decreases inhibitory noncovalent interactions of activation loop amino acids with catalytic residues, which increases catalytic efficiency. In summary, these two types of changes in the activation loop lead to more access by substrates and enhanced catalysis of bound substrates.
Additional regulation of the kinase occurs through the PH domain which adds additional conditions on Akt conformational changes and subsequent activity. The PH domain "appears to lock the kinase in an inactive conformation and the kinase domain disrupts the phospholipid binding site of the PH domain".
Figure \(\PageIndex{38}\) shows an interactive iCn3D model of the separate AKT Pleckstrin Homology (PH) domain bound to the inner member through just the head group of PIP3 (inositol (1,3,4,5)-tetrakisphosphate) (1unq). Waters H bonded to the ligands is not shown.
-tetrakisphosphate%25C2%25A0.png?revision=1&size=bestfit&width=231&height=263)
 Figure \(\PageIndex{38}\): AKT Pleckstrin Homology (PH) domain bound to inositol (1,3,4,5)-tetrakisphosphate (1unq) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...h9LwZyV9WrJsN6
Figure \(\PageIndex{38}\): AKT Pleckstrin Homology (PH) domain bound to inositol (1,3,4,5)-tetrakisphosphate (1unq) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...h9LwZyV9WrJsN6