
28.14: Programmed Cell Death
- Page ID
- 15123


Introduction
We have discussed often how cell signaling might go awry and lead to cancer. However, there are signaling systems that lead to cell death. There are many ways in which cells can die. We'll discuss not "accidental" cell death but one, apoptosis, that is programmed into the genome and highly regulated. Figure \(\PageIndex{1}\) shows how normal cell proliferation and growth can be modulated by two classes of genes, oncogenes that cause proliferation, and tumor suppressor genes that inhibit it.

Apoptosis involves chromatin aggregation and cleavage, the concentration of cell material, and apoptosis body formation. Mutations to aberrantly activate oncogenes or inhibit the expression of tumor suppressor genes lead to cancer. These cells would ideally undergo programmed cell death or apoptosis. As with control of proliferation, some genes promote apoptosis as well as anti-apoptotic genes which inhibit programmed cell death. Dysregulation of these can also cause cancer. Apoptosis is an important mechanism to kill viral-infected cells. However, this can go too far. For example, T helper cells (TH) infected with the HIV virus die. However, the collapse of the population of these cells is in part attributed to apoptosis.
As we learn more about programmed cell death, it is clear that apoptosis is not the only way the genome is programmed to cause cell death. These other ways include:
autophagy - This is a catabolic pathway in which intracellular proteins, protein complexes, and organelles are collected into large autophagosomes in which incorporated lysosomes and their degradative enzymes reprocess damaged or unneeded cell material. It is a highly programmed process, which if dysregulated, could lead to cell death.
necroptosis: Infections and toxins are known to cause necrosis, which is a "passive" form of cell death. In contrast, programmed necrosis is called necroptosis.
Overview of apoptosis.
Apoptosis consists of 4 steps:
- the decision to activate the pathway;
- the actual "suicide" of the cell;
- engulfment of the cell remains by specialized immune cells called phagocytes;
- degradation of the engulfed cell.
The actual steps in cell death require:
- condensing the cell nucleus and breaking it into pieces
- condensing and fragmenting of cytoplasm into membrane-bound apoptotic bodies; and
- breaking chromosomes into fragments containing multiple numbers of nucleosomes (a nucleosome ladder)
To commit suicide must be an extremely important cellular decision. Hence you would expect this process to be regulated and highly complicated. When would it be advantageous to the organism to want a cell to kill itself (or be told to kill itself)? Cell death would be used to:
- "sculpt" an organism during development such as during embryo development, metamorphosis, and tissue atrophy
- regulate the total number of cells.
- defend and remove unwanted or dangerous cells like tumor cells, virally infected cells, or immune cells that recognize self (which could lead to autoimmune disease).
Unregulated apoptosis could exacerbate or cause diseases such as:
- AIDS, in which T helper cell numbers plummet. Part of the dramatic decline in these cells might be caused by health T helper cells being tricked into committing suicide;
- neurodegenerative diseases like Alzheimer's;
- ischemic stroke, when restricted blood flow to certain regions of the brain can lead to neural death through increased apoptosis'
- cancer, in which tumor cells lose their ability to undergo apoptosis;
- autoimmune disease, in which self-reactive immune cells trick normal body cells to kill themselves;
- viral disease;
Apoptosis does not require new transcription or translation, suggesting that the molecular machinery required for cell death lay dormant in the cell, and just requires appropriate activation. What "signals" induce apoptosis?
Signals can be extracellular:
- a hormone (such as thyroxine that causes apoptosis in tadpole tails
- a lack of a "survival" signal (which inhibits apoptosis) such as a growth factor
- a cell:cell contact from an adjacent cell
Signals can be intracellular:
- ionizing radiation
- virus infection
- oxidative damage from free radicals
Apoptosis
Much of this section was derived directly from the following reference, with modifications and additions.
Fox, J., MacFarlane, M. Targeting cell death signaling in cancer: minimizing ‘Collateral damage’. Br J Cancer 115, 5–11 (2016). https://doi.org/10.1038/bjc.2016.111. Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
There are two apoptotic pathways in cells:
- The extrinsic pathway: extracellular apoptotic ligands bind to membrane death receptors, leading to the assembly of the death-inducing signaling complex (DISC). Similar to the inflammatory response we have already discussed in Chapter 5, two specific cysteine-aspartic proteases (caspases) are activated, caspases 8 and 10. These activate other caspases in an amplification of the process.
- The intrinsic pathway: intracellular signals such as damaged DNA or proteins are sensed by Bcl-2 proteins on the outer membrane of mitochondria. The BcL-2 (B-cell lymphoma-2) family of proteins all have Bcl homology domains. Their functions are carried out at the outer mitochondrial membrane. Some members of this family are antiapoptotic (Bcl-2, Bcl-xL, Mcl-1, Bcl-w, A1/Bfl-1, and Bcl-B/Bcl2L10), while others are proapoptotic (Bid, Bim, Puma, Noxa, Bad, Bmf, Hrk, Bik Bax, Bak, and Bok/Mtd). Apoptosis leads to activation of Bax/Bak, which initiate mitochondria degradation, starting with damage to the outer membrane and release of pro-apoptotic proteins like the inner membrane space protein cytochrome C into the cytoplasm. This leads to the assembly of the apoptosome and activation of caspases 9 and 13. Again this is very similar to the formation and activity of the inflammasome which we saw in Chapter 5.
An overview of the extrinsic and intrinsic apoptotic pathways is shown in Figure \(\PageIndex{2}\). We will explore some of the proteins involved in the section below.

The extrinsic death receptor pathway is activated by death receptor ligands, including FasL, TNF-α, DR3, DR4, and DR5 or TRAIL, etc. FasL is an integral membrane protein found in cells. In addition, there are soluble versions of it. The binding of FasL to Fas, an integral membrane protein, initiates the recruitment of FADD, TRADD, and caspase-8 to form the DISC complex, which in turn activates caspase-8 and downstream caspases. The binding of tissue necrosis factor alpha (TNF-α) to its receptor, TNFR1 (a Fas protein), initiates the recruitment of TRADD, RIP, TRAF2/5, and cIAP1/2 to form complex I, which activates NF-κB and JNK pathways and increases the transcription of pro-survival genes. However, the modification of RIP or degradation of cIAP1/2 can lead to the disassociation of complex I. TRADD and RIP then associate with FADD and caspase-8 to form complex II, the so-called death complex.
The intrinsic death receptor pathway is initiated by the BH3-only protein, BCL-2 homology 3 (BH3-only), under intracellular stress such as DNA damage. The BH3-only proteins activate apoptosis by binding and neutralizing the pro-survival proteins, allowing Bax/Bak to homo-oligomerize and permeabilize the mitochondria. For example, BH3-only protein can inactivate Bcl-2 and prevent Bcl-2 from effectively neutralizing Bax and Bak, leading to the activation of Bax and Bak. The activated Bax and Bak on the mitochondrial membrane alter its permeability, depolarizes the membrane, and leads to the release of cytochrome c and Smac, normally found in the inner membrane space, from mitochondria. Figure \(\PageIndex{3}\) shows how monomeric BAK can form an altered dimeric form in the presence of detergent.

The extended left-hand helix on the right-hand side is color-coded to show nonpolar residue (orange) and the polar/charged amino acids in gray. That same section of the protein is shown in cyan in the monomeric protein to the right. One can easily imagine how the apparent amphiphilic helices of the BAK dimer could bind to the outer mitochondrial membrane and alter its structure.
Cytoplasmic cytochrome c associates with Apaf-1 and caspase-9 to form the apoptosome, which activates caspase-9 and downstream executing caspases. Smac can regulate apoptosis by inhibiting the inhibitor of apoptosis proteins (IAPs)." Zhou and Li. Chapter 9, Apoptosis in Polycystic Kidney Disease: From Pathogenesis to Treatment. License: This open-access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0)
Another diagram of the extrinsic and intrinsic apoptotic pathways that show more detail on the domain structures of some key protein and the "executioner" caspases is shown in Figure \(\PageIndex{4}\).
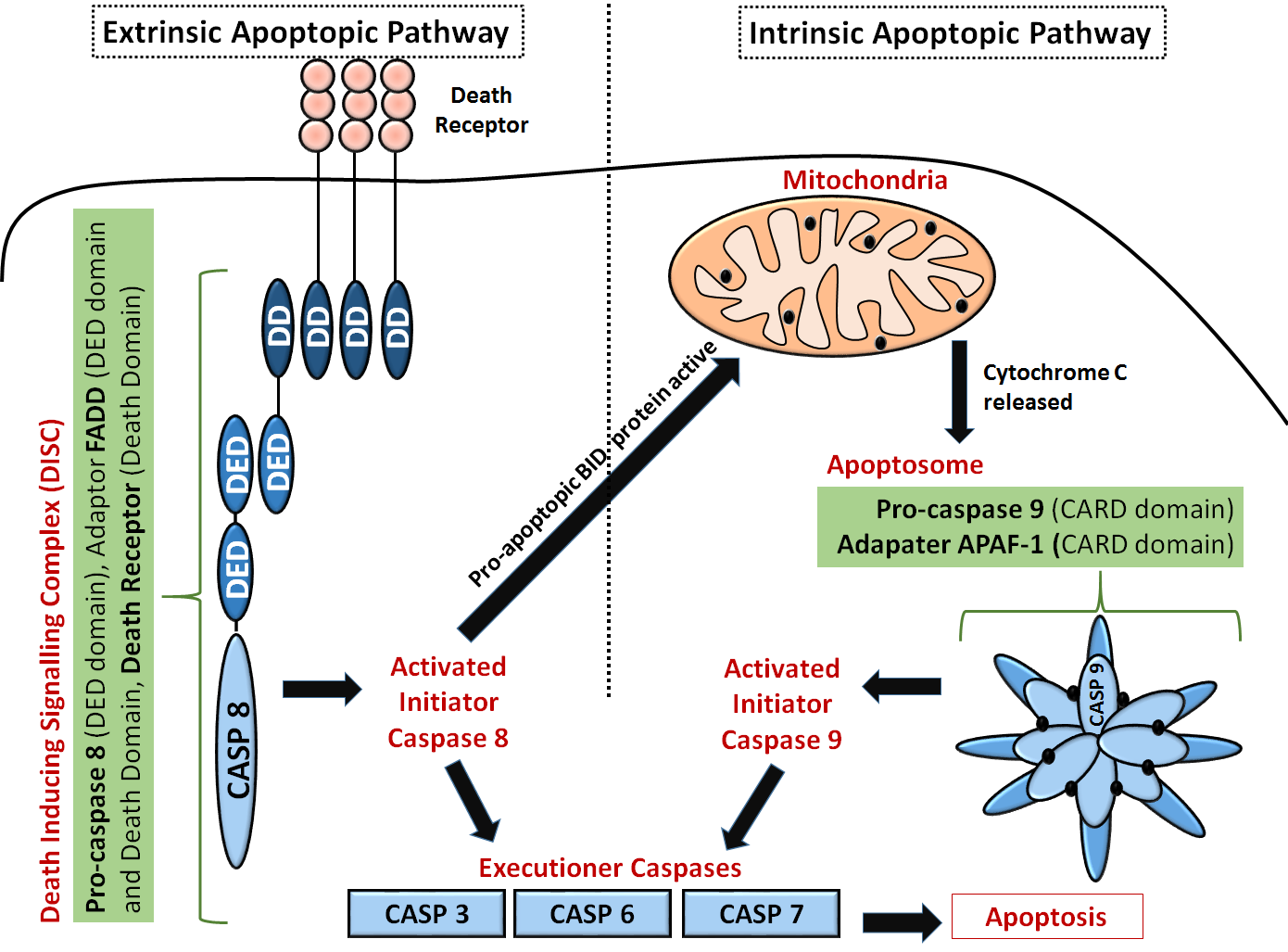
Extrinsic pathway: The first step is an association of death receptors with their cognate ligands, which leads to the recruitment of adaptor molecules, including FAS-associated death domain protein (FADD), and then caspase 8. Caspase 8 cleaves and activate caspase 3 and caspase 7 and can proteolytically activate BH3-only protein BH3-interacting domain death agonist (BID). Proteolytically activated BID (tBID) promotes mitochondrial membrane permeabilization through the activation of the assembly of BAX-BAK channels and represents the main link between the extrinsic and intrinsic pathways.
Now let's look more closely at the ligands that activate the extrinsic pathway, as shown in Figure \(\PageIndex{5}\).

Soluble Fas and soluble FasL bind to the respective ligands inhibiting activation of the pathway. FLIP inhibits the activation of caspase-8 and is thus a major anti-apoptotic protein. Volpe E et al. (2016) Fas–Fas Ligand: Checkpoint of T Cell Functions in Multiple Sclerosis. Front. Immunol. 7:382. doi: 10.3389/fimmu.2016.00382. Creative Commons Attribution License (CCBY).
Now we are in a position to examine the actual structure of some key components of the extrinsic pathway.
Active human apoptosome with procaspase-9 (5JUY)
Figure \(\PageIndex{6}\) shows an interactive iCn3D model of the active human apoptosome with procaspase-9 (5JUY)
.png?revision=1&size=bestfit&width=465&height=425)
Each of the 7 different subunits of the apoptotic protease-activating factors (Apaf-1) is shown in a different color. The seven yellow subunits are cytochrome Cs. The 4 red subunits underneath the disk plane of the other subunits are the zymogen procaspase 9s. The small spacefill CPK color ligands are 2'-deoxyadenosine 5'-triphosphate. The Apaf-1:pc9 pairs, interacting through their CARD domains, form a spiral underneath the disk.
Apaf-1 can be considered an adaptor protein with an N-terminal caspase activation and recruitment domain (CARD), followed by a nucleotide-binding and oligomerization domain (NOD, also known as NB-ARC).
Figure \(\PageIndex{7}\) shows the domain structure of caspase 9 and Apaf-1.
Caspase 9 domain structure 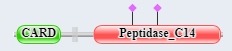 |
Apaf-1 |
Figure \(\PageIndex{7}\): domain structure of caspase 9 and Apaf-1.
The presence of CARD domains in both allows their mutual binding and the assembly of the full apoptosome.
An AlphaFold model of the Cas 9 zymogen
Figure \(\PageIndex{8}\) shows an interactive iCn3D model of human Cas 9 AlphaFold model (P55211)
2.png?revision=1)
The green is the CARD domain and the salmon is the caspase (peptidase_C14) domain. Procaspase 9 is cleaved at Asp 315 (sticks, CPK colors, labeled) into two chains for activation. The activated Cas 9 has two key active site residues, His 237 and the catalytic nucleophile C287 (sticks, CPK colors, labeled). Phosphorylation at Thr-125 by MAPK1/ERK2 blocks procaspase activation by proteolysis. to block caspase-9 processing
Apaf-1
Oligomeric Apaf-1 mediates the cytochrome c-dependent autocatalytic activation of pro-caspase-9 (Apaf-3), leading to the activation of caspase-3 and apoptosis
Figure \(\PageIndex{9}\) shows an interactive iCn3D model of Human Apaf-1 AlphaFold model (O14727)
.png?revision=1)
Domain colors:
- The green is the N-terminal CARD domain
- light red NB-ARC (nucleotide-binding and oligomerization domain - NOD)
- purple is Apaf
- yellow is WD40, gold the C-terminal WD40.
Again the model above does not show the actual structure since the intrinsically disordered regions are not more structured.
The CARD domain of Cas 9 inhibits the catalytic domain of Cas 9. When the CARD domain of Cas 9 interacts with the CARD domain of Apaf-1, the autoinhibition is removed. In addition, the Apaf-1 stimulates the catalytic activity of the protease.
Before assembly into the apoptosome, Apaf-1 is monomeric and in an inactive dATP or ATP conformation. When cytochrome C is released into the cytoplasm, it binds to the WD domains, facilitating a dATP/ATP-cleavage associated conformation change in the Apaf-1, which in the presence of heat shock protein 70 (Hsp) folds to form which leads to the assembly of the active apoptosome.
Fas - Tumor necrosis factor receptor superfamily member 6 - P25445
Figure \(\PageIndex{10}\) shows an interactive iCn3D model of Fas-Tumor necrosis factor receptor superfamily member 6 AlphaFold model (P25445)
.png?revision=1)
The green is the N-terminal TNFR/NGFR domain that is highly enriched in Cys (spacefill, color CPK) in disulfide bonds. The gray spheres are the transmembrane helix. The Red shows the Death Domain.
The death domains are common protein:protein binding domains that serve as adaptors or scaffolds. They can form homo- or heterodimers with other proteins containing the domain which is a part of the CARD domain, DED (Death Effector Domain), and PYRIN.
Human FasL and a soluble Fas Receptor DcR
Figure \(\PageIndex{11}\) shows an interactive iCn3D model of the complex of Human FasL and Its Decoy Fas Receptor DcR (4MSV)
.png?revision=1&size=bestfit&width=334&height=378)
The three gray subunits are soluble decoy receptor (DcR) versions of the Fas TNFR/NGFR domain, which again is highly enriched in Cys. It is structurally very similar to its typical membrane receptor ligand Fas (tumor necrosis factor receptor superfamily member 6 - P25445)
DcR is a secreted member of the TNF family and disrupts apoptosis, which can allow tumors to survive.
Fas and FADD death domain interactions
Figure \(\PageIndex{12}\) shows an interactive iCn3D model of two Fas death domains bound to two FADD death domains (3EZQ)
tetrameric arrangement of four FADD death domains bound to four Fas death domain
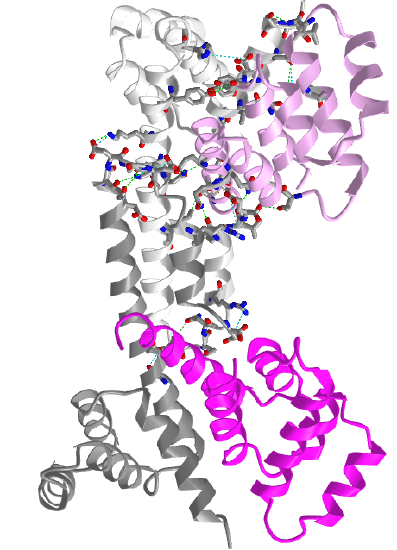
Two Fas death domains are shown in a different shade of gray. The two FADD death domains are shown in a different shade of magenta. Each domain consists of six alpha-helical bundles. Interaction between the dark and light grays Fas death domains and between the light gray Fas and light magenta FADD death domains are shown with side chains in stick with CPK colors.
The Fas-FADD-procaspase 8 complex is collectively called the Death Inducing Signaling Complex (DISC). The Fas-FADD interactions lead to the binding of capase 8 and the completion of the DISC. The actual disc appears to contain 4 FADD death domains bound to 4 Fas death domains. Conformational changes allow both FADD:Fas and Fas:Fas interactions, some of which are weak but when formed switch on the activity of the building complex. The need for 4 monomers probably prevents accidental assembly which would be deadly to the cell.
Mechanism and regulation of apoptosis
Caspases
Characterization of apoptotic mechanisms and cellular players started with the study of C. elegans, a roundworm. The mature worm has about 1000 cells. During development, 131 cells die. Two mutations were found in which the 131 cells did not die. These mutations were called ced3 and ced4 (ced stands for cell death). The sequence of ced 3 was very homologous to a protein called interleukin converting enzyme (ICE) which is required for proteolytic activation of the precursor to interleukin 1, a protein hormone released by certain immune cells during activation and which can promote inflammation. This suggested that proteolysis was required for apoptosis. Subsequent studies show that a whole family of proteases (about 10 in humans) called caspases (ICE has been renamed caspase 1) are required for programmed cell death. These proteases are found in the cell in an inactive form which must undergo limited proteolysis for activation. These caspases form a cascade of proteases which are activated in this process. They are endoproteases that have an active site Cys (C) and cleave at the C-terminal side of Asp residues (asp) and hence are known as caspases - cys containing-asp specific proteases).
ICE is not normally involved in apoptosis, but its artificial activation in cultured mammalian cells can lead to it. Each caspase had the same sequence as they are designed to cleave, so it became evident that they probably cleave each other in an activation cascade mechanism, similar to the coagulation protease cascade of activation of precursors (zymogens) of serine proteases which activate the next in the series. Two series of caspases seem to be involved. One set initiates the process of caspase activation. Just as in the clotting system, the question of what activates the first caspase appeared problematic until investigators found that the initiator caspase can be activated if they aggregate to a critical concentration. This could occur by binding a suicide signal molecule at the cell surface. Conformational changes in the receptor can lead to aggregation of surface receptor molecules with concomitant aggregation of intracellular caspases which interact with the aggregated receptors.
Intracellular signals
How might intracellular activators of apoptosis (like radiation or reactive oxygen species) work? Research indicated the involvement of mitochondria in the apoptotic pathway. Believe it or not, cytochrome C, the heme protein which acts as a water-soluble mobile carrier of electrons in mitochondrial oxidative phosphorylation, shuttling electrons through cytochrome C oxidase or complex IV, leaks out of the intermembrane space and binds to a cytoplasmic protein called Apaf-1 for apoptotic protease activating factor-1. This then activates an initiator caspase-9 in the cytoplasm.
These proteins seem to leak out of mitochondria after a collapse of the electrochemical potential across the inner membrane. The potential collapses as a consequence of the opening of a channel called a nonspecific inner membrane permeability transition pore, composed of both an inner membrane protein (adenine nucleotide translocator - ant) and an outer membrane protein (porin, the voltage-gated anion channel - VDAC). These proteins act together, probably at sites where the inner and outer membranes are in contact. This channel passes anything smaller than molecular weight 1500. Collapsing the proton gradient uncouples oxidation and phosphorylation in the mitochondria. Changes in ionic strength cause a swelling of the matrix. Since the inner membrane is highly convoluted and has a much greater surface area than the outer membrane, swelling of the matrix leads to a rupture of the outer membrane, spilling the inner membrane space proteins (cytochrome C and Apaf-1) into the cytoplasm.
What causes all these changes in the mitochondria? Several interrelated events appear to be involved:
- disruption of ox-phos. and electron transport, caused by irradiation and certain second messengers such as ceramide.
- changes in cell redox potential and generation of reactive oxygen species (ROS).
- DNA damage (caused by radiation, ROS, etc). A protein called p53 is often expressed in cells with DNA damage. Expression of this protein results in inhibition of cell division, or apoptosis, both of which would keep the damaged cell from becoming a tumor cell. Hence the p53 gene is a tumor suppressor gene. It is inactivated by mutation in approximately 50% of all human tumor cells studied. p53 can induce gene expression. Of the 14 different genes whose expression is significantly altered by p53, many seem to be used by cells to generate or respond to oxidative stress. Cells undergo p53 apoptosis through oxidative damage.
- increases in intracellular calcium ions through signal transduction.
Caspase targets:
Apoptosis involves:
- condensing of the cell nucleus and breaking into pieces
- condensing and fragmenting of cytoplasm into membrane-bound apoptotic bodies
- breaking chromosomes into fragments containing multiple numbers of nucleosomes (a nucleosome ladder)
How does caspase activation lead to these events? A protein has been uncovered that when cleaved by a caspase leads to a nuclear breakup. The target protein is usually bound to another protein, a DNA endonuclease. When the target protein is cleaved, the DNase is free to migrate to the nucleus and begin the execution. Membrane changes in apoptosis occur when caspase 3 cleaves gelsolin, a protein involved in maintaining cell morphology. The cleaved gelsolin cleaves actin filaments inside the cell. Another protein is necessary to form apoptotic bodies: a kinase named p21-activated kinase 2 (PAK-2). This kinase is activated by caspase-3 by limited proteolysis. Caspases also cleave beta-amyloid precursor protein which might generate more beta-amyloid protein, causing neural cell death in Alzheimer patients.
Controlling Apoptosis
It should be clear that cells keep tight control of the caspases. Two players which appear to inhibit apoptosis are the mitochondrial proteins Bcl-2 and Bcl-X, which can block the release of cytochrome C from the mitochondria. The Bcl family of proteins has a hydrophobic tail and binds to the outside surface of mitochondria and other organelles like the nucleus and endoplasmic reticulum. These proteins seem to be able to form ion channels in liposomes. So far 15 members of this family (related to ced-9 of C. elegans) have been discovered in humans. Bcl-2 can also bind to Apaf-1 (mentioned above) and inhibit its activation of initiator caspase-9. Bcl-2 is regulated by changes in the expression of the Bcl-2 gene, by post-translational phosphorylation by kinases, or by cleavage by caspases. Overexpression of Bcl-2 can cause a cell to become a tumor cell. Other members of the family, BAX and BAD bind to mitochondria and facilitate apoptosis by stimulating cytochrome C release.
In addition, other proteins called IAPs (inhibitors of apoptosis) can inhibit caspase or other apoptotic proteins. Some viruses make the protease to keep their host cells viable.
Cell Membrane Events
Cells can be instructed to undergo apoptosis through cell surface interactions with other cells which are often immune cells. One of the jobs of the immune cell is to destroy an altered cell (for example a virally-infected cell or a tumor cell). Immune cells themselves must also die after they are activated in an immune response. Activated lymphocytes (like cytotoxic T cells or natural killer cells) can target and kill cells using several ways which can involve apoptosis. In one, an activated lymphocyte binds to a target cell (like a virally infected cell) and secretes perforin, a protein that assembles in the target cell membrane to form a transmembrane channel. Other proteins released by the activated lymphocyte can enter the target cell through the pore and initiate apoptosis. One such protein that enters, granzyme B is a protease that activates caspases in the target cell.
Target cells that express a specific membrane protein called CD95 (also called Fas) are also targeted for apoptosis. This protein receptor, a member of the tumor necrosis factor receptor (TNFR) binds to a membrane protein-ligand on the surface of an activated lymphocyte called CD95 Ligand - CD95L- (also called the Fas ligand). On binding, the CD95 (Fas) receptors on the target membrane aggregate after conformation changes. An adapter protein in the cell, FADD (Fas-associated death domain) binds to the aggregated cytoplasmic domain (the death domain) of CD95 (Fas) and recruits inactive caspase-8 to the site, where their concentration increases. This leads to the activation of the caspases.
This mechanism is used to get rid of activated lymphocytes after they have finished their work. Activated immune cells start expressing Fas a few days after activation, targeting them for elimination. Some cells which have been stressed express both Fas and Fas ligands and kill themselves. Various cells express CD95 (Fas), but CD95L (Fas-Ligand) is expressed predominately by activated T cells.
Cell surface events also can inhibit apoptosis. The binding of "survival" factors (like growth factors) to cell surface receptors can shut off apoptotic pathways in the cells. Some survival factor receptors are coupled to PI-3-kinase (phosphoinositol-3-kinase) through the G protein ras (p21) which is targeted to the cell membrane by post-translational addition of a hydrophobic anchor. The activated kinase produces PI-3,4-P2 and PI-3,4,5-P3, which activates Akt, a Ser/Thr protein kinase. This activated kinase phosphorylates the proapoptotic-protein BAD, which then becomes inactive. In addition, active Akt phosphorylates procaspase, which in its phosphorylated form will not interact with cytochrome C, hence inhibiting apoptosis.
The endpoint of apoptosis is the engulfment of the fragmented cell by a phagocytic cell (such as a macrophage). In a recent article (Nature, 405, pg 85, 2000), it was shown that the activity of phagocytes could be inhibited stereospecifically by the addition of phosphatidyl serine (PS) to the mixture, but not by other negative phospholipids. If you remember from our description of lipids, PS is found exclusively in the inner leaflet of red blood cells). The investigators cloned a gene from the phagocytic cell for a receptor that recognizes PS. When added to ordinary T and B lymphocytes (immune cells), these cells could also take up apoptotic cells. The gene is homologous to genes in Drosophila (fruit fly) and C. elegans (roundworm) suggesting that it is conserved in nature. The message: when cells undergo apoptosis, PS, normally found only in the inner leaflet, is exposed to the outside. It can then bind to receptors on phagocytic cells to complete the process of apoptosis.
Therapeutics
Figure \(\PageIndex{13}\) shows points of therapeutic intervention in the intrinsic and extrinsic apoptotic signaling pathways.

Intrinsic and extrinsic apoptotic signaling pathways and points of therapeutic intervention. Apoptosis can be initiated by signals originating from either the plasma membrane via death receptor ligation (extrinsic pathway) or at the mitochondria (intrinsic pathway). Stimulation of the extrinsic pathway by TRAIL results in TRAIL receptor (TRAIL-R) aggregation and formation of the DISC, in which pro-caspase 8 becomes activated and initiates apoptosis by direct cleavage of downstream effector caspases. The addition of either agonistic TRAIL-R1/R2 antibodies or recombinant human TRAIL (rhTRAIL) has been used to trigger the extrinsic pathway for therapy. The intrinsic pathway is regulated by the BCL-2 family of proteins, which regulate pore formation in the outer mitochondrial membrane and the release of apoptogenic factors such as cytochrome c or SMAC from the mitochondria. The release of cytochrome c into the cytosol triggers caspase 9 activations through the formation of the cytochrome c/Apaf-1/caspase 9-containing apoptosome complex. SMAC promotes caspase activation through the neutralizing the inhibitory effect of IAPs. The intrinsic pathway has been targeted for therapy either by blocking the inhibitory action of the pro-survival BCL-2 family proteins with BH3 mimetics or by inhibiting the anti-apoptotic action of IAPs with SMAC mimetics. The extrinsic and intrinsic pathways are interconnected, for example, by BID, a BH3 domain-containing protein of the BCL-2 family, which upon cleavage by caspase 8 triggers intrinsic apoptosis, thereby further amplifying the signal from the extrinsic pathway.
The entire pathway
Now we can present detailed pathways showing apoptosis in its complexity. Trace the interconnections in the different views.
Figure \(\PageIndex{14}\): View 1

Figure \(\PageIndex{15}\) presents a second view
Pathway 2 https://www.sinobiological.com/pathw...ceptor-pathway
