
5.4: Complementary Interactions between Proteins and Ligands - The Immune System and Immunoglobulins
- Page ID
- 14942


Introduction to the Immune System
Now let's consider the daunting task faced by the immune system - to recognize all possible "foreign" molecules and react to them, either by targeting them for elimination or, paradoxically, to recognize them but not react to them (a process called tolerance). The same can be said of "self-molecules., and the immune system must recognize them but not respond to them, otherwise autoimmune disease might arise in which the body's powerful immune system targets self.
It is virtually impossible to give an in-depth description of the immune system in a short section. Our goal is to illustrate how the immune system recognizes such a vast number of molecules. We will briefly cover the innate and adaptive immune system and their differences, and how some cells (macrophages in particular) in the innate immune system and cells (B and T cells) in the adaptive immune response recognize and respond to target molecules and cells. Finally, we'll discuss how the immune system can respond to similar molecules through the recognition of common molecular patterns. Emphasis will be given to recognition. Ways to simplify the complexities of the immune system have been presented in a fantastic book written by Lauren Sompayrac, How the Immune System Works. (2003, Blackwell Publishing. ISBN: 0-632-04702-X) and adopted here.
We realize we have not yet reached the chapters on carbohydrates, membrane proteins, and nucleic acids. Nevertheless, we present the material in this section to organize it in one specific location. Users can revisit this page after they studied subsequent chapters.
Before we start, think of the variety of chemical species that the immune system should recognize as foreign:
- a bacterial glycan or glycolipid on the outside of the cell
- a viral surface protein, such as the spike protein of the SARS Coronavirus 2
- bacterial dsDNA (and not host dsDNA)
- viral dsRNA (which is not common in host systems
- a self-protein that has been modified in a tumor cell
- a crystal of urea
- extracellular ATP (a place where it is not usually found)
- a silica particle found in particles like asbestos.
How would you design an immune system to bind each of the "enemy" targets above? That is what we will explore in this section - the binding interactions. What happens after the binding is beyond the scope of this section and falls generally in the field of signal transduction - how binding events at the self surface are transferred into intracellular responses.
Three lines of defense protect us from the "enemies", foreign substances (bacteria, viruses, and their associated proteins, carbohydrates, and lipids) collectively called antigens.
- physical barriers of cells that line our outside surface and our respiratory, GI tract, and reproductive systems.
- , the innate immune system (IS) that all animals have. Composed of scavenger cells like macrophages (MΦ), neutrophils, dendritic cells, and natural killer cells (NK) that can move around the body through the blood and lymph systems and burrow into tissues to meet the enemy where they can engulf and destroy bacteria and "cellular debris". Macrophages start as immature circulating monocytes, which enter tissues by slipping through blood vessel walls. They differentiate into macrophages. There they lie in wait ready for the enemy.
- the adaptive immune system, which, as its name implies, can change and adapt to new molecular threats. This branch is better at dealing with viruses, which do their damage inside host cells. The adaptive IS is comprised of B cells that make and secret protein antibodies that recognize specific foreign molecules, and T cells.
In a world experiencing the most deadly pandemic (COVID) of the last 100 years, and with more to come, immune recognition must be an important part of any biochemistry text. This chapter section could be a whole chapter, but we'll leave it as a very long section. Let's start with the adaptive immune system, which we can coopt to make vaccines to the major threats we face.
B Cells and Antibodies
B cells and their differentiated forms (B memory and plasma cells) make antibodies. Antibodies bind to foreign molecules (proteins, glycans, lipids, etc), which might neutralize their effects. For example, an antibody can bind to the hemagglutinin molecule of the influenza virus and prevent its entry into cells. We all are now familiar with the utility of vaccines that create antibodies to recognize the spike protein of the acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Antibodies also bind to foreign cells like bacteria, which signals other host immune proteins and cells to come in for the kill. Antibodies are secreted by B cells, which also have a membrane-bound form of the antibody on their surface. This antibody acts like a receptor that binds antigens and, through a signal transduction process, helps to activate the B cell. Mature B cells (those that have previously seen antigens) can secrete lots of antibodies quickly. Surface and secreted antibodies can recognize and bind to almost any molecule.
There are many forms of antibody, also called immunoglobins (Ig). These include IgA, IgG, IgM, and IgD. We will concentrate on the structure of IgG. It consists of 4 chains (a tetramer) of two light chains and two heavy chains. The light chains form disulfide links with the heavy chains and disulfides also link the heavy chains. Effectively, it's one big protein molecule (about 160 K). Figure \(\PageIndex{1}\) shows a spacefill, secondary structure, and geometric cartoon rendering of a mouse IgG protein (pdb ID 1IGT).

The antibody is shaped like a Y. Foreign molecules (antigens) bind at the end of the top tips of the Y with both chains contributing to antigen binding. The structures of both are dominated by antiparallel beta sheets.
Each chain consists of a single N-terminal variable domain (VL or VH) which participates in antigen recognition. The light chains have an additional constant domain (CL) while the heavy chains have 3 constant domains (CH1-CH3). The constant domains are not involved in antigen recognition, but they are involved in effector functions (such as the binding of other immune molecules like complement proteins) to the antigen-bound antibody heavy chain constant regions. Each of the domains is about 100 amino acids. A cartoon structure with the domain structures is shown in Figure \(\PageIndex{2}\).

Two other features are depicted in the above figure. In each variable region of both light and heavy changes, there are hypervariable regions, which contribute to the unique binding features of a given antibody. The regions are also called complementarity-determining regions (CDRs). Membrane-bound forms of antibodies that serve as "receptor" proteins have additional domains now shown in the figure above. The binding site on the antigen recognized by the antibody is called the epitope. The corresponding binding site on the Y-shaped ends of the antibody that recognized the antigen is called the paratope.
In Figure \(\PageIndex{2}\), you can see that the intact full IgG molecule has 12 variable V and constant C domains called the immunoglobulin domain. Each has about 110 amino acids in length, two layers of β-sheets each with 3-5 antiparallel β-strands with a disulfide bond connecting the two layers.
When we discussed domain structure, we indicated that proteins with multiple binding domains can often be selectively cleaved with protease, with cleaved fragments often retaining binding and other functional properties. The same is true with antibodies. Cleavage with either the proteases pepsin or papain forms fragments with binding activities as illustrated in the figure above. Selected protease digestion was used to clarify structure/function relationships in antibody recognition.
When antibodies targeting different antigens were sequenced, it was clear that much variability was found among antibodies in the variable domains of both the light and heavy chains. In those domains, there were also hypervariable regions. The origin of the variability and hypervariability arise mostly from an extremely large number of gene segments (also exons) in the gene encoding the variable domains. The exons can be spliced together at both the DNA and RNA levels to produce many different DNA/RNA sequences. These are decoded into the variable and hypervariable regions of the light and heavy chain proteins. Somatic mutations are also enhanced in this region.
We mentioned above that both DNA and RNA splicing occur as B cells mature to become antibody-secreting cells (plasma cells). For those who have studied the Central Dogma of Biology, splicing for primary RNA transcripts should come as no surprise. What is surprising is that the DNA genome of B cells changes on their maturation due to the splicing of multiple exons within the variable chain genes to produce unique coding sequences for each clone of a given B cell. There are sets of exons (V, D, and J) or segments within the genes for the variable chain. As the immune cells terminally differentiate, a unique combination of a VDJ segment forms in the DNA genome, so each terminally differentiated B cell is different. When needed (i.e. when their unique antigen binds to membrane forms of the antibody), the cell secretes a monoclonal antibody.
Figure \(\PageIndex{3}\) shows how the different segments become linked in the DNA and how they can be uniquely spiced in the RNA to produce a unique, monoclonal antibody.

The first antibodies produced by the immune system are often of low affinity. Over time, high affinity (low KD) antibodies are produced. What differentiates high and low affinity binding at the molecular level? Do high-affinity interactions have lots of intramolecular H-bonds, and salt bridges (ion-ion interactions), or are hydrophobic interactions most important? Crystal structures of many antibody-protein complexes were determined to study the basis of affinity maturation of antibody molecules. Clones of antibody-producing cells with higher affinity are selected through binding and clonal expansion of these cells. Investigators studied the crystal structure of four different antibodies that bound to the same site or epitope on the protein antigen lysozyme. Increased affinity was correlated with increased buried apolar surface area and not with increased numbers of H bonds or salt bridges as described in Table \(\PageIndex{1}\) below.
| Antibody | H26-HEL | H63-HEL | H10-HEL | H8-HEL |
|---|---|---|---|---|
| Kd (nM) | 7.14 | 3.60 | 0.313 | 0.200 |
| Intermolecular Interactions | ||||
| H bonds | 24 | 25 | 20 | 23 |
| VDW contacts | 159 | 144 | 134 | 153 |
| salt bridges | 1 | 1 | 1 | 1 |
| Buried Surface Area | ||||
| ΔASURF (A2) | 1,812 | 1,825 | 1,824 | 1,872 |
| ΔASURF-polar (A2) | 1,149 | 1,101 | 1,075 | 1,052 |
| ΔASURF-apolar (A2) | 663 | 724 | 749 | 820 |
Table \(\PageIndex{1}\): Characteristics of Antibody:Hen Egg Lysozyme Complexes (HEL). Data from Y. et al. Nature: Structural Biology. 6, pg 484 (2003)
Many crystal structures of antibody:antigen complexes have been determined. Especially interesting are those in which the antigen is a protein. It is important to understand antibody:protein antigen interactions to develop vaccines against key epitopes in proteins such as the spike protein of the SARS-CoV-2. Let's look in more detail at the antibody that binds to hen egg white lysozyme (HEWL). The crystal structures of many different IgG antibodies that bind HEWL are known. One recognizes a discontinuous epitope on lysosome consisting of the following amino acids: H15, G16, Y20, R21, T89, N93, K96, K97, I98, S100, D101, G102, W63, R73, and L75. Most of these amino acids are polar, and five are charged.
Figure \(\PageIndex{4}\) shows the interaction of part of the Fab fragment of an antibody that binds to the HEWL epitope just mentioned (3hfm). The light chain is shown in magenta, the heavy chain in dark blue, and the antigen lysozyme in gray. The side chains of the amino acids in the epitope of HEWL are shown in sticks. Note the complete complementary of HEWL and Fab surfaces. Water is excluded from the interface.

Figure \(\PageIndex{5}\) shows an interactive iCn3D model of the same HEWL:Fab complex (3hfm). Lysozyme is shown in black.
.png?revision=1&size=bestfit&width=174&height=263)
Here is an external link to an interactive iCn3D model showing a detailed view of the multiple interactions (salt bridges, hydrogen bonds, pi-cation)
T Cells
What happens if a virus makes it into a cell? Antibodies can not bind to them anymore to prevent their entry. Something must be able to recognize a virally infected cell and eliminate it. What about a cancer cell? Wouldn't it be nice if something could recognize a tumor cell as foreign and eliminate it before it divides too much and metastasizes? Those "somethings" are T cells. There are many T cells in a person, and many different kinds, including T helper cells (Th), cytotoxic lymphocytes (CTL), and even suppressor T cells. They express different subsets of proteins that differentiate them and their functions.
T cells also recognize antigens but unlike B cells, these antigens can only be protein fragments. The membrane proteins that recognize protein fragments are called T-cell receptors. In addition, they don't recognize protein antigens in isolation. They must be bound to a protein on the surface of an "antigen" presenting cell (such as a macrophage or dendritic cell). The T cell receptor recognizes and binds simultaneously to the foreign protein fragment and to the self "antigen-presenting" protein on the surface of the antigen-presenting cells. The self-protein that binds and presents the foreign protein fragments (peptides) is called a Major Histocompatibility Complex (MHC) protein.
Antigen-presenting cells like macrophages and dendritic cells have MHC Class II molecules on their surface. These bind protein fragments from engulfed bacteria, for example, and present them on the surface. T cell receptors bind to the peptide:MHC II complex. All cells in the body have MHC Class I proteins on their surface. If a cell is infected with a virus, protein fragments from the virus end up bound to the MHC Class I protein on the surface. Now a T cell can bind through its T cell receptor to the peptide:MHC Class I complex. By displaying a viral protein fragment on the surface, the immune cell can recognize a virally infected cell without getting inside the cell where the virus is. Sompayrac describes MHC molecules as looking like a hot dog bun. In the grove of the bun lies the peptide fragment - like the hot dog. The T cell receptor recognizes both the bun and the hot dog!
Figure \(\PageIndex{6}\) shows an interactive iCn3D model of a MHC Class Class I heavy chain complexed with beta-2-microglobulin with a peptide fragment of the vesicular stomatitis virus nucleoprotein (2VAA).
.png?revision=1&size=bestfit&width=340&height=263)
The T-cell receptor consists of two transmembrane protein chains, alpha and beta, each containing a single variable and constant domain, followed by a transmembrane domain. Hence they are less complicated than an antibody chain. They bind through their extracellular variable domains a peptide fragment bound to a MHC Class I or Class II membrane protein in the target cell.
We described above how an undifferentiated B cell has the potential to produce an incredible diversity of different antibodies from a starting genetic sequence. This occurs through both DNA and RNA splicing. The same processes occur with the alpha and beta chains of T-cell receptors. This is illustrated in Figure \(\PageIndex{7}\). Note that the alpha chains have no D (diversity) coding sequences.

Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy. International Journal of Molecular Sciences 21(7):2378 (2020). DOI: 10.3390/ijms21072378. License CC BY
Figure \(\PageIndex{8}\) shows an interactive iCn3D model of the T-cell receptor alpha and beta chains binding to MHC Class 1 protein with a bound peptide (6rp9). The MHC protein complex consists of the histocompatibility antigen, A-2 alpha chain, and β-2-microglobulin, an 11K subunit of MHC Class I proteins but not Class II MHC proteins. Bound to it is the 9 amino acid cancer/testis antigen 1 (shown in spacefill). The peptide is sandwiched between the MHC protein complex and the T-cell receptor α and β chains.
.png?revision=1&size=bestfit&width=479&height=241)
The actual functional structure in vivo is more complicated. The T cell receptor is found within the much larger T Cell receptor complex (TRC), which contains two copies of the CD3 complex, which itself consists of γ, δ, ε, and ζ chains, as shown in Figure \(\PageIndex{9}\) (A). Part A shows the variable and C domains of the α and β chains of the T-cell receptor in green and dark. The rest of the T-cell receptor complex includes two copies each of the CD3 complex, which consist of one copy of εδζ chains and one copy of εγζ chains.

Figure \(\PageIndex{9}\): T cell receptor structor. Kumaresan Pappanaicken R., da Silva Thiago Aparecido, Kontoyiannis Dimitrios P. Methods of Controlling Invasive Fungal Infections Using CD8+ T Cells.
Frontiers in Immunology, 8, 1939 (2018). https://www.frontiersin.org/article/...mmu.2017.01939. DOI=10.3389/fimmu.2017.01939. Creative Commons Attribution License (CC BY).
As mentioned earlier, one of the functions of the MHC Class I molecules is to present peptides derived from tumor antigens to T-cells, which leads to the activation of other immune cells and hopefully destruction of the tumor cells displaying the tumor antigen. Much work has gone into the study of immune surveillance and the ultimate destruction of tumor cells with the hopes of improving our own immune response to cancer cells. In early work, T-cells that had infiltrated tumors were isolated from a patient, amplified in the lab by adding a cytokine (ex interleukin 2 - IL2), a protein growth factor released by activated immune cells, and then re-infusing the tumor-specific T cells along with IL2 back into the patients. This adoptive cell transfer (ACT) therapy led to remissions in some patients but the therapy also could be lethal.
One promising type of immune therapy is chimeric antigen receptor (CAR) T cell therapy or (CAR T) in which patients are treated with modified versions of their own T-cells. T cells are removed from a cancer patient's own blood. A gene is constructed to mimic the V and C domains of the alpha and beta chain of the T-cell receptor and inserted into the patient's T-cell using a viral vector. The gene construct contains as their tumor antigen binding motif the V and C domain of an antibody gene made to recognize the tumor antigen. The receptor is hence a chimeric (formed from parts of different proteins) antigen receptor (CAR) with antibody and T cell parts. The genetically modified cells are amplified and re-infused back into the patient. Once the collected T cells have been engineered to express the antigen-specific CAR, they are "expanded" in the laboratory into the hundreds of millions. This approach is obviously very expensive so some recent successes in using a vius to add the appropriate gene construct without having to remove cells cells from the patients are noteworthy.
Compare the structure of the chimeric antigen receptor (CAR) in Figure XX-C with normal T-cell receptors shown in A. The CAR contains two, single-chain variable fragments (scFv) derived from combining the variable domains of the light (VL) and heavy (VH) chains of an antibody recognizing the tumor cell. These domains are connected by a linker peptide (10-25 amino acids) enriched in glycines, which confers flexibility and serines/threonines for hydrogen bonding interactions. This is attached to an FC fragment and other intracellular effector domains to create the receptor. Figure \(\PageIndex{10}\) shows the scFv structure. We'll discuss the addition cytoplasmic CD28 domain in a bit.

You can imagine this whole T-cell receptor complex involved in the binding of a tumor peptide antigen presented by a MHC I transmembrane protein on a tumor cell, as illustrated in Figure \(\PageIndex{11}\) (in different colors). The entire interacting structure is called the T cell immunological synapse.
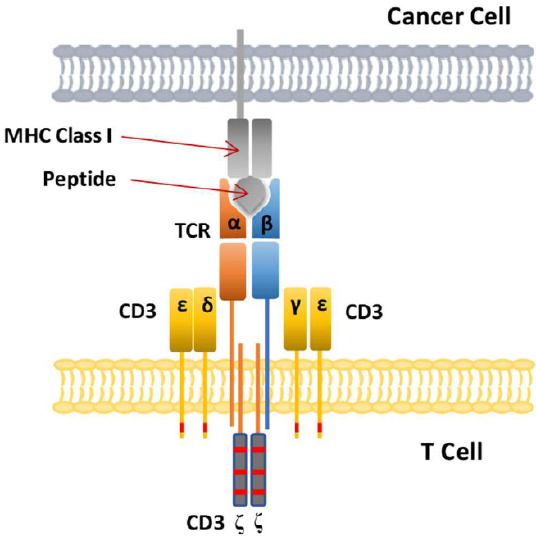
Figure \(\PageIndex{11}\): T cell immunological synapse of T cell with a cancer cell. Zhao Lijun, Cao Yu J. Engineered T Cell Therapy for Cancer in the Clinic. Frontiers in Immunology, 10, 2250 (2019) https://www.frontiersin.org/article/...mmu.2019.02250. Creative Commons Attribution License (CC BY).
Protection Against Autoimmune Recognition - Coreceptors
How can the immune system recognize and bind to any foreign molecule but not self? The subject of immune tolerance is too specialized to include here, but there are a few features we will discuss.
The MHC Class I proteins do present "self" peptides in their binding pockets. Self-proteins are also cleaved into peptides in the cell by proteasomes. However, the T cell receptor does not recognize and bind to the self-peptide fragment bound to the MHC Class 1 protein. Hence T cells do not recognize self and turn against their own cells. Once and a while they do, however, and autoimmune diseases like MS, rheumatoid arthritis, and lupus result.
B cells and T cells must be activated before they can carry out their function. It is important to regulate the "on" switch. If the cells were activated without need, they might turn against self. In addition to T cell receptor complex binding to foreign peptide:MHC complexes for immune cell activation, they must also bind yet another protein on the antigen-presenting cell.
In the case of T helper cells. the T cell protein CD28 must also bind the B7 protein on an antigen-presenting cell like a macrophage expressing an MHC II protein:foreign peptide complex. Hence there is one specific signal (the peptide:MHC complex binding to the T cell receptor complex) and a nonspecific signal (B7 binding CD28). Why are two signals needed for activation? Again, Sompayrac has a great analogy. A safety deposit box at a bank takes two keys, a specific key (which you have) and a "nonspecific" key (which the bank uses for all boxes) to open the box. Think of it as double security. You don't want to activate immune cells for killing unless you need to do so.
Yet other proteins are involved to ensure correct T-cell activations. We'll consider T-cells expressing either the proteins CD4 or CD8. T-cells expressing these expand after antigen stimulation (infection or immunization). It depends on the subtype of T-cell. Let's consider two here:
T cells expressing the protein CD4: After initial simulation, they differentiate and proliferate into the T helper cells named TH1, if they produce the cytokine interferon (IFN)-γ and TH2, if they produce the cytokine IL4. CD4 is an integral membrane protein and acts as a co-receptor for MHC Class II:peptide complex found on cells like macrophages. These "present" foreign antigens and antigens that are part of microbes taken up into phagosomes in antigen-presenting cells like macrophages. Hence they are involved in the protection of phagosomal spaces similar to the role of antibodies which bind to and protect from antigens in extracellular spaces. Traditional vaccines targeted to foreign proteins such as the spike protein of the SARS-Cov 2 virus mainly elicit antibodies that bind to antigens in the blood and other intracellular spaces. Such vaccines are not very effective against antigens such as the malaria parasite in blood since the parasite stays in circulation only an hour before it is internalized in liver cells.
T-cells expressing CD8: These cells produce cytokines (IFN-γ and tumor necrosis factor (TNF)-α) or secrete protein which form pore-forming complexes on foreign cells, leading to their lysis of cells such as pathogens or tumor cells. The CD8 protein has an alpha and beta subunit. They serve as co-receptors for MHC Class I:peptide complex found on tumor cells for example. MHC Class I proteins are found on most cells of the body (see below). Cytotoxic T-cells (a type of T-cell) express CD8. Vaccines that elicit a robust CD8 response might be more effective in the defense against intracellular pathogens that cause malaria, tuberculosis, and acquired immune deficiency syndrome (AIDS). Memory CD8 cells protect against intracellular infections so more effort is being made to develop CD8 T-cell vaccines.
Figure \(\PageIndex{12}\) shows the multiple co-signals that are required to activate the CD4 T-cell (blue sphere), which has the T-cell receptor complex, the co-receptor CD4, and the CD28 protein. It also displays a cytokine receptor, which binds cytokines released by the antigen-presenting cells (macrophage shown in pink). This leads to the proliferation and differentiation of the activated T-cell.

Sompayrac asks another interesting question. Why is antigen presentation by MHC proteins necessary at all? B cells don't really need presentation since they can bind antigen with membrane antibody molecules. Why do T cells need it? He gives different reasons for Class I and Class II presentations:
Class I MHC (found on most body cells): T cells need to be able to "see" what is going on inside the cell. When virally infected cells bind foreign peptide fragments and present them on the surface, they can be "seen" by the appropriate T cell. It's a way to get a part of the virus, for example, to the surface. They can't hide out in the cell. T cells don't need to recognize extracellular threats since antibodies from B cells can do that. Presentation is also important since viral protein fragments found outside of the cell might bind to the outer surface of a noninfected cell, targeting them for killing by the immune system. That wouldn't be good. It also helps that peptide fragments are presented on the surface. This allows parts of the protein that are buried and not exposed on the surface, which would be hidden from interaction with outside antibodies, to be used in signaling infection of the cell by a virus.
MHC Class II (found on antigen-presenting cells like macrophages): Two different cells (the presenting cell and the T helper cell) must interact for a signal for immune system activation to be delivered to the body. Again it is a safety mechanism to prevent the nonspecific activation of immune cells. Also, as in the case above, since fragments are presented, more of the foreign "protein" can contribute to the signal to activate the immune system.
Recognition and Response in the Innate Immune System
The B and T cell part of the immune system represents the more sophisticated branch of the immune system called the adaptive immune system. It can be trained to recognize any foreign chemical/cellular species. The other branch of the immune system is the innate immune system. The system recognizes common molecular structures found all many different organisms, so in this branch, there is no need to adapt to each foreign species individually. The adaptive immune response also must be activated by cells of the innate immune system.
The innate immune system recognizes common structural features in viruses and living cells like bacteria, fungi, and protozoans like amoebas. The cells of the innate system (dendritic cells, macrophages, eosinophils, etc, which we talked about as antigen-presenting calls above) have receptors called Toll-like Receptors 1-10 (TLRs) that recognize the common pathogen-associated molecular patterns (PAMPs), which leads to binding, engulfment, signal transduction, maturation (differentiation), antigen presentation, and cytokine/chemokine release from these cells. Dendritic cells, which reside in the peripheral tissues and act as sentinels, are an example. They can bind PAMPs which include:
- CHO/Lipids on bacteria surface (LPS)
- mannose (CHO found in abundance on bacteria,
- yeast dsRNA (from viruses)
- nonmethylated CpG motifs in bacterial DNA
After entering an immune cell, bacterial and viral nucleic acids are recognized by intracellular TLRs. Dendritic cells phagocytize microbial and host cells killed through programmed cell death (apoptosis). During maturation, surface protein expression is altered, allowing the cells to leave the peripheral tissue and migrate to the lymph nodes where they activate T cells through the antigen presentation methods described above. They also control lymphocyte movement through the release of chemokines. Figure \(\PageIndex{13}\) shows the TLR family, their binding signals, and intracellular adapter proteins used to transmit signals into the cell.

Figure \(\PageIndex{14}\) shows an interactive iCn3D model of the mouse Toll-like receptor 3 ectodomain (that sticks out into the cytoplasmic space from an internal organelle) complexed with double-stranded RNA (3CIY).
.png?revision=1&size=bestfit&width=421&height=239)
Double-stranded RNA is found in the life cycle of many viruses so it makes great sense for evolution to create a binding protein to recognize this common structure (PAMP). The TLR3 ectodomains (ECDs) form dimers when the dsRNA is at least 40-50 nucleotides long. The dsRNA is shown in spacefill (cyan and magenta). Note the extensive glycosylation (colored cubes) in the structure. The protein looks like a "horseshoe-shaped solenoid " with lots of beta structure. It has 23 leucine-rich repeats (LRRs) with some conserved asparagines allowing for extensive hydrogen bonding. One face appears to be free of carbohydrate residues and may be important in dimerization and function.
Messenger RNA vaccines against the SARS-Cov2 spike protein have probably saved up to 20 million lives in the first year of the COVID-19 pandemic. The development of mRNA vaccines will go down as a great scientific achievement that requires decades of fundamental and applied research by many scientists.
Vaccines usually are composed of target proteins from a virus, for example. Instead of delivering an actual protein, whose actual development and mass production takes years, why not use mRNA that encodes a viral protein or fragments of it? The idea has been around for a long time. The problem is that RNAs, with their 2'-OH on the ribose ring, are very labile and degrade easily. In addition, injecting RNA into a patient causes a significant immune response to the RNA. We mount immune responses to foreign viral RNA through our TLR receptors (TL3, 7, and 8) but what is needed is an immune response to the protein made from the injected mRNA, not to the RNA. Yet we don't make an immune response against our own RNA. Why?
Two major problems had to be solved (and a host of others as well) to make mRNA vaccines, the problem of stability and our immune response against them. A hint comes from the observation that TLRs recognize non- or undermethylated DNA found in bacteria. Methylated CpG motifs in DNA do not stimulate an immune response. Katalin Karikó, Michael Buckstein, Houping Ni, and Drew Weissman reported in 2005 that the incorporation of methylated (m) and modified nucleosides m5C, m6A, m5U, s2U, and pseudouridine ablated the immune response to the RNA. This opened the door to mRNA vaccines. The paper was rejected by Nature and Science but published in Immunity (Vol. 23, 165–175, August 2005. DOI 10.1016/j.immuni.2005.06.008). The last line from the paper was truly prophetic: "Insights gained from this study could advance our understanding of autoimmune diseases where nucleic acids play a prominent role in the pathogenesis, determine a role for nucleoside modifications in viral RNA, and give future directions into the design of therapeutic RNAs".
Katalin Karikó and Drew Weissman were awarded the 2021 Lasker–DeBakey Clinical Medical Research Award (often a prelude to the Nobel Prize) for their fundamental research that has saved so many of us. They were awarded the Nobel Prize in Medicine in 2023. See Chapter 9.1 for more details about the role of pseudouridine in mRNA vaccines.
Inflammasome
Think of the things you would want your immune system to protect you from. Of course, there are the pathogens like viruses, bacteria, and fungi. And of course, you want to be protected from yourself in that you don't want to activate your immune system with self-antigens. But what about "non-biological" molecules like silica or asbestos whose presence might be deleterious? What about normal biomolecules (proteins, nucleic acids) that suddenly find themselves in the wrong cellular location due to cell death by necrosis or physical injury?
In the previous section, we discussed how innate system immune cells (dendritic cells, macrophages, eosinophils, etc) have receptors that recognize common pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharides (LPS) on the surface of bacteria, mannose on bacteria and yeast, flagellin from bacterial flagella, dsRNA (from viruses) and nonmethylated CpG motifs in bacterial DNA. These antigens are recognized by pattern recognition receptors (PRRs) - specifically the Toll-like Receptors (TLRs) 1-10. These include plasma membrane TLRs (TL4 for LPS, TL5 for flagellin, TLR 1, 2, and 6 for membrane and wall components of fungi and bacteria) and intracellular endosomal TLRs (TLR3 for dsRNA, TLR 7 and 8 for ssRNA and TLR9 for dsDNA)
Damage-associated molecular patterns (DAMPs) are typically found on molecules released from the cell or intracellular compartments on cellular damage (hence the name DAMP). Many are nuclear or cytoplasmic proteins released from the cells. These would now find themselves in a more oxidizing environment which would further change their properties. Common DAMP proteins include heat shock proteins, histones and high mobility group proteins (both nuclear), and cytoskeletal proteins. Think about what non-protein molecules might be released from damaged cells that might pose problems. Here are some other common non-protein DAMPS: ATP, uric acid, heparin sulfate, DNA, and cholesterol crystals. In the wrong location, these can be considered danger signals.
If TLRs recognize PAMPs, what recognizes DAMPs? They are recognized by another type of intracellular pattern recognition receptor (PRR) called NOD (Nucleotide binding Oligomerization Domain (NOD)- Like Receptors or NLRs. NLRs also recognize PAMPs. The proteins also are named as the Nucleotide-binding domain (NBD) and Leucine-Rich repeat (LRR)–containing proteins (NLR)s. This family of proteins participates in the formation of a large protein structure called the inflammasome. (Sorry about the multiple abbreviations and naming systems!)
As both PAMPs and DAMPs pose dangers, it would make sense that once they recognize their cognate PRRs (TLRs and NLRs, respectively), pathways leading from the occupied receptors might converge in a common effector system for the release of inflammatory cytokines from immune cells. Given that uncontrolled immune effector release from cells in an inflammatory response might be dangerous, it would be sometimes helpful to require two signals to trigger cytokine release from the cell. We've seen this two-signal requirement for the activation of T cells.
Two such inflammatory cytokines are Interleukin 1-beta (IL 1-b) and IL-18. Activation of TLRs by a PAMP leads to activation of a potent immune cell transcription factor, NF-kbeta, which leads to transcription of the gene for the precursor of the cytokine, pro-interleukin 1-beta. Without a specific proteolytic cleavage, the active cytokine will not be released from the cell.
The protease required for this cleavage is activated by a signal arising when a DAMP activates a NLR, which then through a sequence of interactions leads to the proteolytic activation of another inactive protease, procaspase 1, on a large multi-protein complex called the inflammasome. (In later chapters we will see other such protein complexes with targeted activities - including the spliceosome, which splices RNA to produce mRNA, and the proteasome which conducts controlled intracellular proteolysis). The activated inflammasome activates procaspase to produce the active protein caspase (a cysteine-aspartic protease).
The convergence of the signals from the PAMP activation of a TLR and DAMP activation of a NLD at the inflammasome is shown in Figure \(\PageIndex{15}\).
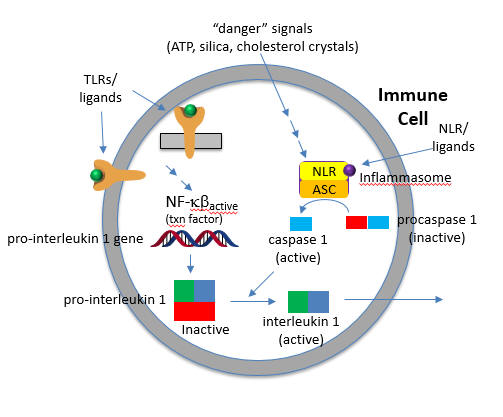
The active cytokine interleukin 1-beta helps recruit innate immune cells to the site of infection. It also affects the activity of immune cells in the adaptive immune response (T and B cels). Active IL-18 leads to the increase of another cytokine, interferon-gamma and it also increases the activity of T cells that kill other cells.
The focus of this chapter is on binding interaction and their biological consequences. From that perspective, this section will address
- the structure and activity of caspases, which activate the pro-cytokine prointerleukin 1 beta,
- the structure and ligands for the NLRs, the structure and properties of the inflammasome, and finally
- how "danger" molecules such as ATP and crystals (cholesterol, silica) activate the inflammasome.
Unfortunately, there are many proteins involved with crazy acronyms for names. These proteins have multiple domains and many of the proteins often have multiple names. Sorry in advance!
Caspases
Caspases (Cys-asp-proteases), not to be confused with Cas9 (CRISPR associated protein 9, an RNA-guided DNA endonuclease) is a protease which when active can lead to cell death, or in a less austere fashion initiate the inflammatory response (sometimes good, often bad or even fatal). They have an active site nucleophilic Cys and cleave peptide bonds after an Asp in target proteins. All caspases (13 in humans) have an N-terminal pro-domain followed by large and small protease catalytic domain subunits. As with other proteases, it is found as an inactive zymogen. Why is this important?
To become activated they are recruited to a scaffolding protein where they are activated by removal of the N-terminal domain of the zymogen and then a second cut between the large and small catalytic subunits. The enzyme that does this is caspase itself in an autocatalytic step. There are 3 kinds of caspases, two of which are involved in programmed cell death. We'll discuss the inflammatory cytokine processing of Caspase-1. Once activated, the initiators activate other effector (executioner) caspases in the cell). Caspase 1 is activated by the inflammasome.
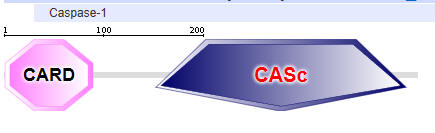
Two major domains are found in Caspase 1, the caspase recruitment domain (CARD) which mediates self-interaction with scaffold and adaptor proteins in the inflammasome for activation, and a proteolytic catalytic domain, as shown in Figure \(\PageIndex{16}\). All domain structures in the section were obtained using Conserved Domains from the NCBI (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) or the Simple Modular Architecture Research Tool (SMART) at the EMBL ( http://smart.embl-heidelberg.de/smart/set_mode.cgi?NORMAL=1 ). Uniprot was used for protein (FASTA) sequences (http://www.uniprot.org/uniprot/). We will see the CARD domain often.

NOD-like receptor proteins (NLRPs)
The NOD-like receptor proteins (NLRPs) are a family of proteins with similar domain structures. The structures and abbreviations used for the molecular players in inflammasome activation are very complicated and confusing. Different programs show different domains, which adds to the complexity. We will attempt to reduce the confusion by just showing domain structure diagrams, even if some show different domains for the same protein. Remember domains are calculated from structure so different algorithms using different databases might return different domain structures. Table \(\PageIndex{2}\) below shows the domain structure for NLRPs.
| NLRP1,2,3 | 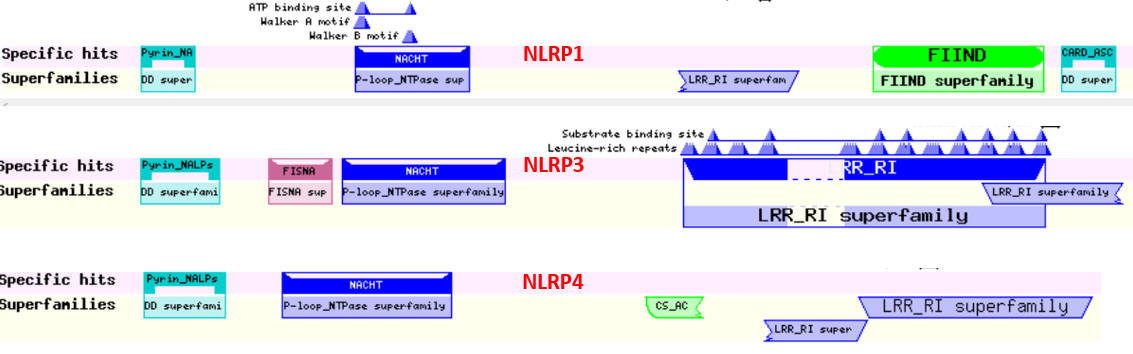 |
| NLRP1 (NALP1) | 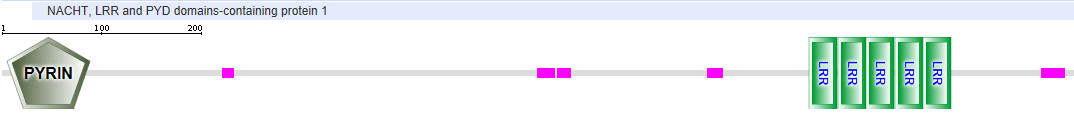 |
| NLRP3 (NALP3) | 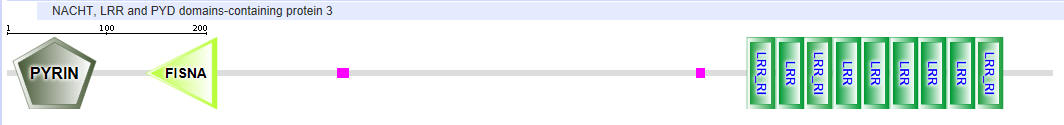 |
| NLRP4 | 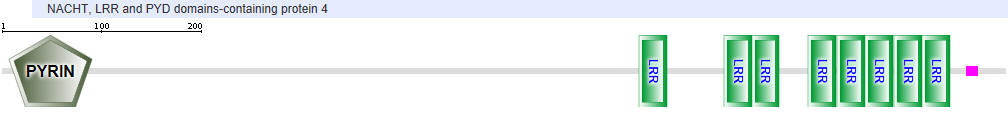 |
| NAIP1 |
|
| NAIP2 |
|

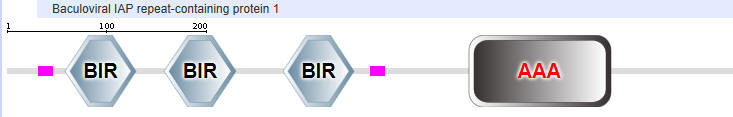

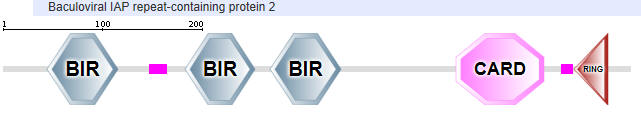
Table \(\PageIndex{2}\): Domain structures of the NLRPs
Another protein in the NLR family is NAIP (neuronal apoptosis inhibitor protein). The domain structure for NAIP1 is shown in Table \(\PageIndex{2}\) above. In contrast to the other NLR for which specific ligands have not yet been found, several NAIPs have been shown to bind specific PAMPs. NAIP1 binds the needle protein CprI from C.violaceum which starts to drive the assembly of the NLRC4 inflammasome. NAIP2 binds the inner rod protein of the bacterial type III secretion system (which for Salmonella typhimurium is the protein PrgJ). NAIP5 and NAIP6 bind bacterial flagellin (which for Salmonella typhimurium is the protein FliC). AAA in the second domain representation is ATP-associated activities in the cell (otherwise denoted as the NACHT domain in the top representation).
NAIP2 interacts with another adapter NLR family protein, NLRC4 (NLR family CARD domain-containing protein), to form the inflammasome. The domain structure of NAIP2 is shown in Figure \(\PageIndex{x}\) below:
Note that many of these proteins share common domains:
- Pyrin-NALP - Pyrin domains on different proteins self-associate through inter-protein Pyrin:Pryin interactions
- NACHT - This domain contains about 300-400 amino acids and can bind ATP and may cleave it (i.e. act as an ATPase)
- LRR - for Leucine Rich Repeat. These 20-30 amino acid repeats may occur up to 45 times in a given protein. They fold into an arc shape and seem to facilitate protein:protein interactions. On the concave side of the arc, they have a parallel beta sheet while on the convex side, they have an alpha helix. They also appear to be involved in the binding of PAMPS and DAMPs;
- CARD - for caspase activation and recruitment. CARD domains on different proteins self-associate through inter-protein CARD:CARD interactions;
- BIR - Baculoviral inhibition of apoptosis protein repeat;
- ASC - Apoptosis-associated speck-like protein containing a CARD Adapter domain, allowing it to interact with other proteins with a CARD domain.
ASC Adaptor Protein
Small adapter proteins like ASC with a CARD domain mediate the binding of caspases in the apoptosome (involved in apoptosis or programmed cell death) and in the inflammasome. This smaller protein has two domains, a pyrin domain and a CARD domain as shown in Figure \(\PageIndex{17}\). It is required for the recruitment of caspase-1 to some inflammasomes (for example, ones that contain NLRP2 and NLRP3

The Active Inflammasome
The active inflammasome, in general, consists of three different kinds of proteins, some present in multiple copies: NLRPs, adapter proteins like ASC, and procaspases. They may also contain additional recruitment and ligand sensor proteins. We'll discuss two types using different NLRPs, the NLRP4 and NLRP3 inflammasome.
NLRP4 Inflammasome
Some of the best structures (obtained by cryomicroscopy) are for the NAIP2:NLRP4 inflammasome. Figure \(\PageIndex{18}\) shows part of the complex consisting of 11 NLRP4 subunits arranged in a large ring. The actual biological complex has 1 NAIP2 subunit and 10 NLRP4s.

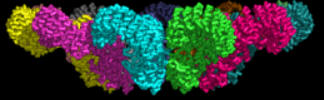
Figure \(\PageIndex{19}\) shows an interactive iCn3D model of the activated NAIP2-NLRC4 inflammasome (3JBL))
.png?revision=1)
Figure \(\PageIndex{19}\): Activated NAIP2-NLRC4 inflammasome (3JBL). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...8Ae7vY4aX1SLX8
How does this structure arise? Presumably, it would not exist in the absence of a PAMP or DAMP to minimize immune-mediated inflammatory damage. Data suggests that the bacterial protein PrgJ (denoted PrgX in the figure below) binds to its receptor, NAIP2, altering its conformation as shown in Figure \(\PageIndex{20}\). This binary complex presents an asymmetric electrostatic surface which allows a loose association with NLRP4, which leads, after a conformational change, to a tighter binding interaction. Nine more NLRP4s bind similarly to form the 11-subunit ring structure.
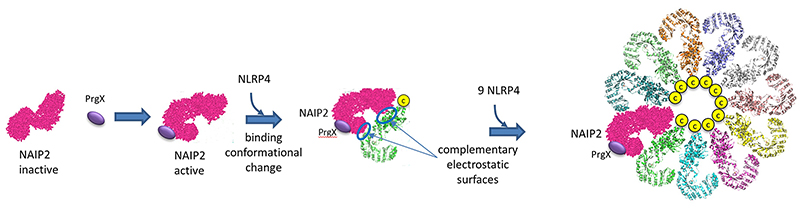
The complementary electrostatic interactions between two of the many NLRC4 subunit monomers in the NLRP4 inflammasome are depicted in Figure \(\PageIndex{21}\).
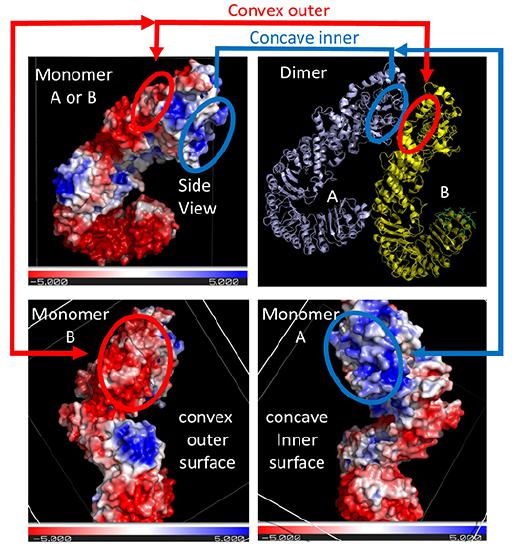
The top right panel shows two of the NLRC 4monomers (of the 12 in the Jsmol model) bound to each other, with the concave inner face of the A subunit interacting with the convex outer face of the B subunit. For simplicity, only the interactions at the top of the dimers are highlighted. The other panels show the electrostatic potential surface (red indicating negative and blue positive) for each monomer on a sliding scale of -5 to +5 (images created using the PDB2PQR Server and Pymol).
The depicted negative (red) electrostatic potential outer surface on one NLRC4 monomer that is complementary to the positive (inner) surface on the other NLRC4 subunit is outlined in each panel in red or blue ellipses, respectively. The curved tertiary structure of the proteins and the opposing electrostatic surface potentials of opposite faces commit the subunits to form a large ringed 12-mer core of the nucleosome.
Note that this assembled ring brings together the CARD (caspase recruitment domain, yellow circles/spheres) which can interact with the CARD domain of the procaspase protein through CARD:CARD inter-protein interactions (think of a stack of playing cards all stuck together in a deck of cards) as shown in Figure \(\PageIndex{22}\).
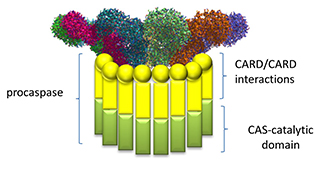
Once assembled, proximal procaspases autocatalytically convert procaspase 1 into active caspase 1, which can activate, by proteolysis, the cytokines interleukin 1 beta and interleukin 18 to form active cytokines which are released from the cell. Remember that the procytokines are present only if their genes have been transcribed following activation of the transcription factor NF-kappa beta through PAMP binding to a TLR.
NLRP3 Inflammasomes
Figure \(\PageIndex{23}\) shows an interactive iCn3D model of the NLRP3 double-ring cage, 6-fold (12-mer) (7LFH)
_(7LFH).png?revision=1)
Figure \(\PageIndex{23}\): NLRP3 double-ring cage, 6-fold (12-mer) (7LFH). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?DEbdkUoBtqRQ9bu59
The full-length mouse NLRP3 consists of 12- to 16-mer organized in a double-ring cage. It is held together by interactions between the leucine-rich repeats (LRR) domains. The pyrin domains are shielded by the structure, so they will not be activated without appropriate signals. The complex is also localized to the membrane.
In contrast to NLRP4 inflammasomes, which require specific PAMPs/DAMPs for activation, NLRP3 inflammasomes seem to be activated by cellular distress as well as cell exposure to pathogens. It is one of the main responders to a variety of microbial infections. Given the large number of microbes that lead to NLRP3 inflammasome activation, it has been suggested that the actual signal that triggers NLRP3 is indirect. One such indirect signal is K+ ion levels in cells.
In normal cells, K+ ions are higher in the cytoplasm than in the outside of the cell. Potassium ion decreases in cells caused by efflux can activate NLRP3 inflammasomes. Other conditions include the rupture of lysosomes (perhaps associated with the cellular uptake of particles like silica, uric acid, cholesterol crystals, and other "nanoparticles"), altered mitochondrial metabolism (which can lead to reactive oxygen species within the cell), etc. Obviously, all of these danger triggers don't bind to NLPR3 but somehow lead to downstream activation of it. NLRP3 hence probably works by being a general sensor for cell stress.
Inappropriate and chronic activation of inflammation has been associated with many diseases such as cancer, cardiovascular disease, diabetes, and autoimmune diseases. Given the multiple types of signals that can activate the NLRP3 inflammasome, this complex is the focus for active drug development to find inhibitors that would stop undesired inflammation. These inflammasomes are found in granulocytes, monocytes (macrophages), megakaryocytes, and dendritic cells.
Activated NLRP3 recruits the ASC Adaptor Protein, which leads to the recruitment and activation of procaspase 1. NLRP3 has a pyrin, NACHT, and LRR domain. ASC has a pyrin and CARD domain. Active LRP3 can then recruit ASC through pyrin:pyrin inter-protein domain interactions. This then allows the CARD domain of bound ASC to recruit procaspase through CARD:CARD interactions (remember that procaspase has a CARD domain as well), forming the active NLRP3 inflammasome. An added feature of NLRP3 inflammasome activation occurs when the transcription factor NFkb, activated by PAMPs (signal 1), leads to the transcription of both the procytokines (IL-1 beta and IL 18) and of NLRP3 itself.
Hence two signals are again needed:
Signal 1
The first signals are the bacterial and viral (influenza virus, poliovirus, enterovirus, rhinovirus, human respiratory syncytial virus, etc) PAMPs, which bind to TLRs and lead to the activation of the NFkb transcription factor. This activates not only the transcription of pro-interleukin 1-beat and interleukin 18 but also the transcription of the NLRP3 sensor itself.
Signal 2
Signal 2 is delivered by PAMPs and DAMPs indirectly to the sensor NLRP3. This leads to the assembly of the inflammasome. These DAMPs appear to prime the activation of NLRP3 protein and subsequent formation of the active NLRP3 inflammasome. But what activates NLR3P3? After many studies, it became clear that the typical bacterial ligands that would activate TLRs and perhaps NLRs only prime NLRP3 for activation. They don't bind to it directly.
Extracellular ATP is a major activator of NLRP3. Nanoparticles are known to release ATP as well. Most studies show that K+ efflux from the cell is an early signal and that the NEK7, a protein that phosphorylates other proteins, binds to NLRP3 after potassium ion efflux and activates it. Removing NEK7 stopped NLRP3 but not NLRP4 inflammasome activation. Although NLRP3 bound to NEK7 through the NEK7 catalytic domain, the activity of the catalytic domain of NEK7 was not needed.
What leads to K+ efflux? Let's back up to possible upstream events that could lead to efflux and try to find a link to ATP. The background for some of this material will be explored in future chapters. The following steps occur as shown in the figure and information below:
- solids such as silica, cholesterol crystals, uric acid crystals, and even aggregated proteins such as prions can be engulfed by monocytes/macrophages (much as they engulf bacteria as part of their immune function) in a process called phagocytosis. The particles are enveloped in plasma bilayer-derived membrane which buds off into the cell. This vesicle merges with a lysosome which gets damaged in the process. They then release ATP into the cytoplasm;
- cytoplasmic ATP can then move outside of the cell through the glycoprotein membrane channel called pannexin 1;
- extracellular ATP can bind to another membrane protein called the P2X7 purinoceptor. This protein now becomes a cation channel which allows K+ efflux since the ion has a higher concentration inside the cell than outside, as illustrated in Figure \(\PageIndex{24}\). The extracellular ATP "gates" open the P2X7 cation channel. The pore-forming toxin nigericin from Streptomyces hygroscopicus also leads to potassium ion efflux. Likewise, pore-forming proteins from S. aureus (hemolysins) lead to potassium ion efflux and activation of the NLRP3 inflammasome. We will discuss membrane protein in great detail in a later chapter.
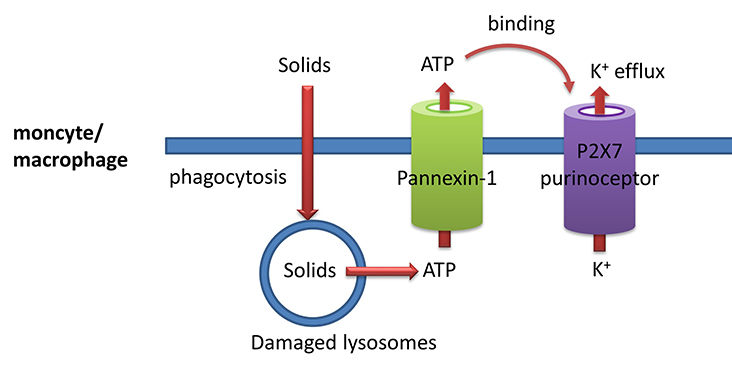
Other signals also activate the NLRP3 inflammasome. These include mitochondrial damage and the release of reactive oxygen species.