
4.8: Protein Folding and Unfolding (Denaturation) - Dynamics
- Page ID
- 14937


Introduction
We've seen many static and rotatable images of lipid aggregates (the micelle) as well as proteins. We have learned some rules about the disposition of amino acid side chains in a folded proteins. However, when we think about how proteins fold, we have to think dynamically as well as thermodynamically. Figure \(\PageIndex{1}\) shows a fun but clearly unrealistic animation of how a protein might fold from an unfolded state with exposed hydrophobic side chains (orange) to a folded state when they are mostly, but not fully, buried.
Luckily we have the tools of molecular dynamics (MD) at our fingertips which helps us imagine how these processes take place and concomitantly how to probe protein folding experimentally.
As protein folding occurs in 3D, let's explore a free energy (G) landscape for folding from an extraordinarily large number of unfolded states of higher free energy to a single low energy folded state. Two such landscapes, created by Ken Dill's group, are shown in Figure \(\PageIndex{2}\).

The right image B shows a simplistic ("much-reduced frustration") funnel view with a simple one-step path for any unfolded protein to reach a single global free energy minimum state, a process that occurs without any intermediates. A more realistic view is shown in A in which there are a series of local minimums and one global minimum. Like in any activation energy curve you have encountered in chemistry courses, traversing from a local minimum to other local minimums or the global minimum is possible if enough energy is provided to overcome the activation energy for that step. Panel A implies that are many intermediates on the road to the final global free energy minimum native state but that local minimum could be populated and either stable or metastable depending on their activation energies. Intermediates might be "trapped" in these local minima. The view also conforms to our view that proteins are conformationally flexible and can adopt a variety of conformations. This is especially true for intrinsically disordered proteins.
Now let's add some additional complexity. A protein on the path to a folded state has more hydrophobic exposure than the native state, so you would expect that it could aggregate with other self proteins and form intermediates and end products off of the normal folding pathway. These are illustrated the Figure \(\PageIndex{3}\) which shows the "normal" protein folding (blue) and a misfolding (orange) landscape. The misfolded landscape is populated with amorphous aggregates, oligomers, protofibrils and fibrils.

We will discuss protein folding done in the lab (in vitro) as well as protein folding in the cell (in vivo). In vitro folding is done in very defined conditions, typically using low concentrations of small proteins to minimize misfolding opportunities. Folding in vivo occurs as a protein is being made on a ribosome. It also occurs when a fully-folded protein misfolds (such as during fevers in disease states) and has been prevented from folding by association with other molecules. Folding in vivo is often assisted by other proteins called folding chaperones.
In either case, given the number of possibly nonnative states, it is amazing that proteins fold to the native state at all, let alone in a reasonable time frame. Consider this greatly simplified view of protein folding for a protein containing 100 amino acids. If each amino acid can adopt only 3 possible conformations, the total number of conformations could be 3100 = 5 x 1047. Assuming that it would take 10-13 s to change each conformation, the time required to "test" all conformations would be 5 x 1034 s or 1027 years, longer than the age of the universe (14 x 109 yr). Yet the protein can fold within seconds. This paradox is called the Levinthal paradox, after Cyrus Levinthal.
Lubert Stryer, (in his classic Biochemistry text), shows a way out of this dilemma by using an analogy of a monkey sitting at a typewriter, and typing this line out of Hamlet: "Me thinks it is like a weasel." Random typing would produce that line after 1040 keystrokes on average, but if the correct letters were maintained, the number of keystrokes would be in the realm of a few thousand. Proteins could fold more quickly if they retain native-like intermediates along the way. Also, remember that much of conformational space is already restricted by allowed phi/psi angles (remember the empty areas in the Ramachandran plots).
We will explore the classic study of the folding of RNase done by Anfinsen, for which he won the Nobel Prize. RNase is a small protein with 4 disulfide bonds and can refold from denaturing and reducing conditions in vitro. If he had chosen a larger protein, he might not have met with success as they are more prone to aggregation. We'll discuss mostly small proteins in this section.
Before we start, let's anticipate what might happen to fully reduced and denatured RNase with 8 free Cys side chains. If the denatured protein is suddenly placed in a refolding solution without denaturant and in an oxidizing environment (such as oxidized glutathione), the reduced cysteine side chains could start forming disulfide pairs, but only one combination of such pairs would be native. What would be the probability that a purely random process of forming disulfides would produce 4 correct ones and a fully native state?
To think about that, try this thought experiment. You have 4 pairs of socks, with each pair having a different color as shown in Figure \(\PageIndex{4}\). You threw them into a drawer unpaired after washing. Now without looking, take one sock out and then a second and tie them together to form a pair. Continue doing this without looking until all four are paired. What is the probability that all the socks will be correctly matched when you are done? Once you have the answer, you're ready to understand Anfinsen's classic experiment. To think about that, try this thought experiment.
You have 4 pairs of socks, with each pair having a different color as shown in Figure \(\PageIndex{4}\). You threw them into a drawer unpaired after washing. Now without looking, take one sock out and then a second and tie them together to form a pair. Continue doing this without looking until all four are paired. What is the probability that all the socks will be correctly matched when you are done? Once you have the answer, you're ready to understand Anfinsen's classic experiment.

- Answer
-
We'll calculate the probability that you get 4 perfect matches. You pick one sock initially. You have a 1/7 chance to get the match from the remaining 7 unpaired socks. You have 6 socks left. Pick one again. From the remaining 5 you have a 1/5 chance of getting a match. Now pick another. From the remaining 3 you have a 1/3 chance of getting a match. Now pick another. You have a 1/1 changes of getting the match. Each is independent so the total probability of getting 4 matched pairs of socks is given by the products of the probability at each step, or
(1/7) x (1/5) x (1/3) x (1/1) = 1/105 or about 1%
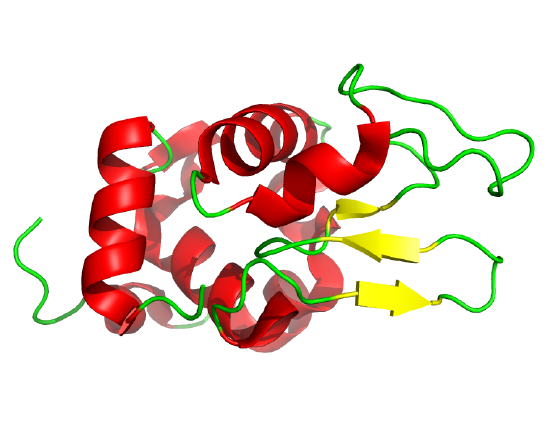
The classic experiment of Anfinsen showed that, at least for some proteins, all the necessary and sufficient information required to direct the folding of a protein into the native state is present in the primary sequence of a protein. Anfinsen studied the in vitro folding of a single chain protein, RNase, which has four intrachain disulfide bonds, as shown in the interactive iCn3D model in Figure \(\PageIndex{5}\).
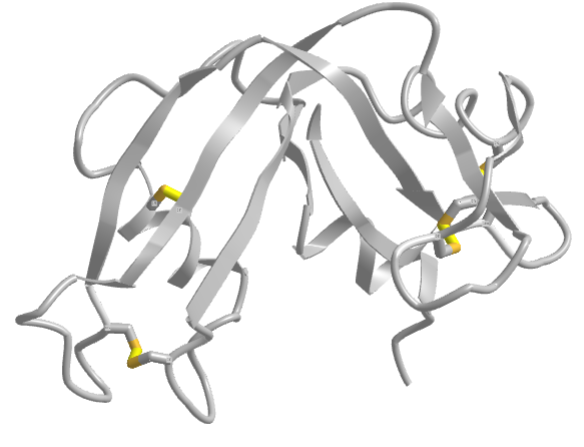
 Figure \(\PageIndex{5}\): RNase with four intrachain disulfide bonds (yellow sticks) (1KF5). (Copyright; author via source).
Figure \(\PageIndex{5}\): RNase with four intrachain disulfide bonds (yellow sticks) (1KF5). (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...Risnvb1SfNrww6
We have previously discussed how chemical agents (such as beta-mercaptoethanol, a disulfide reducing agent) can covalently interact with specific protein functional groups. Some can bind through complementary intermolecular forces to the active site or other cavities on the surface. Other reagents, like urea, acting through generalized solvent changes or nonspecific interactions with the protein, can alter protein folding. Anfinsen used two different reagents, 8 M urea and beta-mercaptoethanol (βME), in combination to unfold, or denature, RNase to the nonnative or denatured state. He then removed the βME using dialysis, allowing the disulfides to reform. Next, he removed the denaturing reagent, urea. To monitor if the protein was correctly refolded or renatured, he tested the activity of the protein compared to the native protein. He found that the "refolded" protein retained only 1% of its initial activity (compare this to the matching socks activity). If, however, he added catalytic amounts of βME, the protein soon retained 100% of its initial activity. For his work, he was awarded the Nobel Prize in Chemistry in 1972. A general outline of his experiment is shown in Figure \(\PageIndex{6}\).

Figure \(\PageIndex{7}\) presents a chemical mechanism to show how catalytic (non-stoichiometric) amounts of beta-mercaptoethanol can lead to full recovery of the most stable set of a protein with two disulfide bonds (right hand side).

Scientists have investigated the folding of proteins both in vitro and in vivo. In vitro experiments involve denaturing the protein with urea, guanidine hydrochloride, or heat, then refolding the protein by removing the perturbant (denaturing agent), using spectral techniques to follow the process. In vivo experiments involve the study of intracellular proteins that assist folding. An understanding of protein folding can not be separated from an understanding of protein stability, and an understanding of the nature of the native and denatured state as illustrated in the protein folding landscapes shown in Figure \(\PageIndex{1}\) and Figure \(\PageIndex{2}\).
In studying protein folding and stability/structure of the native and denatured states, both equilibrium (thermodynamic) and timed (kinetic) measurements are made. Folding occurs in the ms to second range, which limits the ability to study the presence of intermediates in the process. Some clever methods have been developed to study intermediates in protein folding by trapping specific intermediate structures and investigating their structure and stability in a "leisurely" fashion. Alternatively, intermediates can be studied as they occur using stop-flow kinetics. In this technique, a protein under denaturing conditions is rapidly mixed with a solution containing no denaturant or protein by injecting both solutions into a mixer/cuvette using syringes. The denaturant in the protein solution is now diluted such that renaturation can occur. Spectral measurements can begin at once.
A diagram summarizing these methods is shown in Figure \(\PageIndex{8}\). Study it in conjunction with the text which follows.

Folding appears to proceed not by an obligatory pathway but a probabilistic or stochastic search of possible conformations. Evolution has surely selected for sequences that can make it to the folded state. Localized secondary structure motifs (like a short alpha helix and beta turns) can form quickly (about 1 ms). The folding of small proteins occurs, depending on their structure, over a wide time frame (ms to minutes). Most likely, a small number of amino acids coalesce into a core, which serves as a nucleus for folding into structures that are similar to the native state. Finally packing interactions collapse the structure into the native state.
In general, the more complex the fold of the backbone, the longer it takes the protein to fold. If complexity requires more interactions among distal regions of the polypeptide change, then the more complex the fold, the less probable that random interactions would lead to quick protein folding. The mechanisms of folding for larger proteins (greater than 100 amino acids) appear to proceed through intermediates, suggesting that different domains of the protein can fold independently.
Protein Folding In Vitro
Early studies of protein folding involved small proteins which could be denatured and refolded in a reversible fashion. A two state model, D ↔ N, was assumed. The denaturants were heat, urea, or guanidine HCl. Since the denatured states are less compact than the native state, the viscosity of the solution can be used as a measure of denaturation/renaturation. Likewise, the amino acid side chains in the differing states, would be in different environments. The aromatic amino acid Trp, Phe, and Tyr absorb UV light. After excitation, the electrons decay to the ground state through several processes. Some vibrational relaxation occurs, bringing the electrons to lower vibrational energy levels. Some of the electrons can then fall to various vibrational levels at lower principle energy states through a radiative process. The photons emitted are lower in energy and hence longer in wavelength. The emitted light is termed fluorescence. The wavelength of maximum fluorescent intensity and the lifetime of the fluorescence decay is very sensitive to the environment of the amino acids. Hence fluorescence can also be used to measure changes in protein conformation. Other spectral techniques like CD spectroscopy as well as simple absorbance measurements, are used. For small, single domain proteins (such as RNase) undergoing reversible denaturation, graphs showing the extent of denaturation using each technique above, as shown in Figure \(\PageIndex{9}\), are superimposable, giving strong validity to the two state model.

Proteins that fold without easily discernable, long-lived intermediates and follow a simple two-state model, D ↔ N are said to undergo cooperative folding. This simple model needed to be expanded as more proteins were studied. Some intermediates in the process were detected.
Some proteins show two steps, one slow one, and one quick one, in refolding studies, suggesting an intermediate. The longer a protein is kept in the denatured state, the more likely it is to display an intermediate. One accepted explanation for this observation is that during an extended time in the D state, some X-Pro bonds might isomerize from trans to the cis state, to form an intermediate. Alternatively, as in the case of RNase, which has a cis X-Pro bond in the native state, denaturation causes an isomerization to the trans-state. In the case of RNase, to refold, the accumulating intermediate I must reisomerize in a slow step to the cis state, followed by a quick return to the N state.
Some proteins, which contain multiple disulfide bonds that must reform correctly after reductive denaturation could refold into intermediates with the wrong S-S partner. Such intermediates can be trapped by stopping further S-S formation during refolding with the addition of iodoacetamide as shown in Figure \(\PageIndex{10}\).
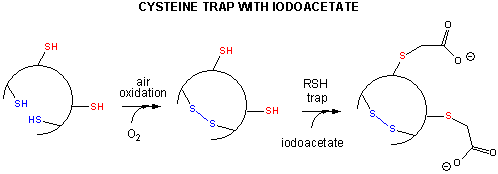
By adding iodoacetamide at varying times along the folding pathway, and separating starting (unfolded), trapped intermediates on the pathway and the final native state by HPLC, the entire folding pathway can in principle be determined. This has been done for many proteins including bovine pancratic trypsin inhibitor (BPTI), small protein with 58 amino acids, a molecular weight of 6512, and three sets of disulfide bonds (C5-C55, C14-C38 and C30-C51). The structure of BPTI with native disulfides is shown in Figure \(\PageIndex{11}\).

Given its characteristics, BPTI has been used as a model protein to study protein folding. These studies have shown that no non-native disulfides form as intermediates in the pathway. Two major intermediates form quickly, each with two of the three native disulfide bonds. These intermediates are named N',with disulfide pairs 14-38 and 30-51, and N*, with pairs 5-55 and 14-38. These two intermediates then slowly form a common intermediate NSHSH, with disulfide pairs 5-15 and 30-51, which then converts very quickly to the native N state with three correct pairs (5-55,14-38 and 30-51). This folding pathway is shown in part a and b of Figure \(\PageIndex{12}\).

In going from N' or N* to NSHSH, the 14-38 disulfide, which is very solvent-exposed (see Figure \(\PageIndex{11}\)), must break before proceeding to the N (native state). Mousa et al replaced that disulfide with a stable methylene thioacetal bridge (MT, alternative name methylenedithioether, S-CH2-S link) with the idea that once this stable (i.e irreversible) 14-S-CH2-S38 bond formed in N' and N*, it would not break again. The formation of the methylenedithioether bond and its properties compared to disulfide bonds are shown in Figure \(\PageIndex{13}\).

Hence the final last intermediate, NSHSH, would not form, leaving just N' and N*. This is illustrated in Part c an d in Figure \(\PageIndex{12}\).
In actuality, MT-BPTI does fold to the native form, whose crystal structure is virtually identical to native BPTI. Figure \(\PageIndex{12}\) shows "2D" folding plots that show HPLC retention time of reactants, intermediates on one axis and folding time on the other for wild-type BPTI (part a) and MT-BPTI (part b). Hence the folding scheme is more complicated than the one shown in Figure \(\PageIndex{14}\).

Some proteins form partially folded but stable intermediates when folded under partially denaturing conditions. A good example is lactalbumin, which under mildly acidic conditions (pH 4), low levels of guanidine HCl, or neutral pH and low ionic strength in the absence of calcium (which normally binds to the protein), forms a stable, isolatable intermediate (I) called the molten globule (MG). Figure \(\PageIndex{15}\) shows the folded state with a bound calcium ion and circular dichroism spectra of the protein in various states.

Data show that the MG is about 50% larger in volume than the N state. This compares to the denatured state, which can be 300% larger than the native state. Hence, it is more like the native state as studied by hydrodynamic techniques, but with more solvent accessibility of hydrophobic side chains. The MG has a similar CD spectrum as the native state, but the aromatic side chains display the same UV absorption and fluorescent characteristics as the protein in 6 M guanidine HCl, suggesting that the final tertiary state has not yet completely formed. The secondary structure in the MG may not be the same as in the native state
NMR can also be used to detect folding intermediates. Using this technique, proteins are unfolded in D2O, which will cause the exchange of all Cs with ionizable protons, including, the amide Hs. An amine is a weak base (pKB around 3.5) so its conjugate acid, the protonated amine, has a pKa of around 9.5. An amide or peptide bond would be a weaker base than an amine since it's lone pair is less available (due to delocalization through resonance) for sharing with a proton. The pKa for the conjugate acid of the amide (in which the amide N is protonated and has a plus charge) is much lower, around -0.5, than the pKa for the conjugate acid of an amine. At 2 pH units greater than its pKa, the charged amide N is close to 100% deprotonated The pka of the protonated group is important since the rate of H exchange is related to the pKa, holding other variables constant. The pka of an unprotonated amine (RNH2 → RNH- is very high (30s) and hence deprotonation of the RNH2 amine to form RNH- is not likely under normal conditions. Figure \(\PageIndex{16}\) shows NMR D/H exchange/protection experiments for the formation of an alpha helix in a protein folding experiment.

Refolding is initiated by diluting the protein into a solution without the denaturatant, but still in D2O. As the protein folds and becomes more compact, the buried atoms are now sequestered from the solvent, and no longer readily exchange Ds. Then the protein is placed in H2O at pH 9.0 for 10 ms, after which the pH is changed to pH 4.0. D → H exchange is promoted at high pH, and quenched for the amide Ds and Hs at low pH. Amide Hs that continue to exchange must be accessible to water. Those that aren't are usually buried in secondary structure.
Hen Egg white lysozyme: Radford et al, Nature 358, pg 303 (1992) 2YVB

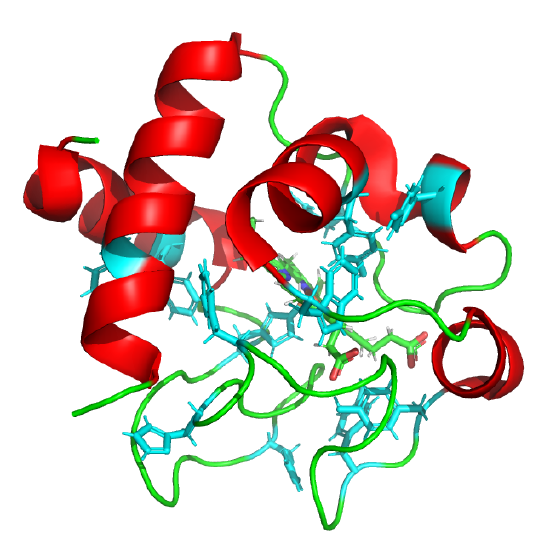
Cytochrome C: Elove et al, Biochemistry, 31, pg 6879 (1992). Cyan aromatic amino acids; Heme not shown. 5TY3

When the same techniques are applied to large, multidomain or oligomeric proteins, only a few percent refold in vitro. Incorrect intermolecular interactions and heterogeneous aggregation appear to be the main problems, which prevent correct protein folding in vitro
Here is a movie of a 6 us molecular dynamics simulation of the small protein villin.
The movie starts with the final crystal structure of villin and show how it folds into its final structure.
With permission from the Beckman Institute for Advanced Science and Technology National Institutes of Health //National Science Foundation, Physics, Computer Science, and Biophysics at University of Illinois at Urbana-Champaign
The Denatured State
Although the structure of native and native-like states can be determined using x-ray crystallography and in solution using NMR, little detailed information exists on the actual structure of denatured and intermediate states. Intermediate states are difficult to trap in a way that allows detailed structural analysis. In contrast to the "native" state which consists of an ensemble of closely related states, intermediates and denatured states would consist of an ensemble of many different states, making structural analysis more difficult. Religa and others from Fersht's lab have engineered a mutant of the engrailed homeodomain (En-HD) from Drosophila melanogaster that allows such structural analyses to be performed. The mutation, Leu16Ala (L16A), destabilizes the protein such that it can be denatured simply by changing ionic strength. It is stable at high ionic strength and folds quickly under those conditions. However, at physiological ionic strength, it is "denatured" but contains significant alpha-helical structure but has nonnative contacts. It behaves like an early folding intermediate in that if placed in solutions of higher ionic strength it rearranges to form the native state. If placed in lower ionic strength, it progressively "unfolds" to yet other states. Given the ambiguities in how to define denatured and early folding intermediates states, Ferscht's group suggests an "explicit" definition of the denatured state. They define the unfolded state (U) as the "maximally unfolded state of a protein, in which the backbone NH groups have little protection against 1H/2H exchange". They define the denatured state, D, as the "lowest energy non-native state under a defined set of conditions". In this scenario, the denatured state could also be a folding intermediate if placed in conditions that promote folding. Previous work from the group showed that the denatured state of En-HD has three helices protected from 2H exchange and was 1kcal/mol l(4.1 kJ/mol) lower in energy than the unfolded state.
In section 4.6, we discussed the added complexities to the notion of a simple 2-state D ↔ N model for protein folding. These include Silent Single nucleotide polymorphisms, Metamorphic Proteins and Intrinsically Disordered Proteins (IDPs).
Protein Folding In Vivo
There are many differences between how a protein might fold or unfold in a cell compared to a test tube.
- The total concentration of all the proteins and nucleic acids in cells is estimated to be about 350 g/L, or 350 mg/ml. Most measurements in the lab are conducted in the range of 0.1 to 10 mg/ml
- Proteins are synthesized in cells from an N to C terminal direction. Hence the nascent protein, as it emerges from its site of synthesis (the ribosome), might fold into intermediate structures since not all of the protein sequence is yet available for direct folding.
- Proteins are synthesized in the cytoplasm, but they have to find their final place in the cell. Some end up in membranes, some must translocate across one or even two different membranes to end up in specific organelles like the Golgi, mitochondria, chloroplasts (in plant cells), nuclei, lysosomes, peroxisomes, etc. Do they translocate in their native state?
Additional evidence suggests that protein folding/translocation requires assistance (i.e. catalysis) in the cell.
- Mutant cells defective in certain proteins can lead to the accumulation in the cells of misfolded and aggregated proteins.
- eukaryotic genes (taken from higher cells, which contain nuclei and internal organelles), when transferred into prokaryotes (bacteria, like E. Coli), can be expressed to form protein, but they often misfold and aggregate in the bacterial cells and form structures called inclusion bodies.
Hence recombinant proteins expressed in vivo have the same problems in folding as larger proteins in vitro. In both cases, conditions favor the accumulation of nonnative proteins with exposed hydrophobic groups leading to aggregation. Aggregation also occurs in vivo when a protein is over-expressed or expressed at a higher temperature than normal. Why? Mutant cells have been selected that actually suppress inclusion bodies in vivo. This effect was mediated by a class of proteins, which are expressed by the bacteria and other cells when their temperature is raised. The function of these proteins, called heat shock proteins (Hsp), was unknown until it was realized that they facilitate correct protein folding, in part, by binding to denatured proteins in the cells before they aggregate into inclusion bodies. Further studies discovered a large number of proteins that seem to facilitate protein folding and prevent aggregation in vivo. These proteins are now called molecular chaperones. Many are still named Hsp#, where # is the approximate molecular weight of the protein in kD on a PAGE gel.
In a broader sense, a protein homeostatic environment exists within cells to maintain the proteome. This system includes chaperones proteins which facilitate folding and refolding, the proteasome, which cleaves undesired proteins marked by ubiquination, and the lysosome, which facilitates the process of autophagy of structures such as damaged mitochondria. Here we will concentrate on chaperones, which also are involved in protein transport in the cell and preventing aggregation. Some have used novel names to describe the various activities of chaperones. These include holdases/translocases, unfoldases/foldases and disaggregases, which are used to process the various potential intermediates and end products that occur. A simplified 2D free energy folding diagram showing the involvement of chaperones is shown in Figure \(\PageIndex{17}\).

Figure \(\PageIndex{17}\): 2D Free Energy Folding Diagram with Chaperone Assist. Moseck et al. Front. Mol. Biosci., 14 June 2021 | https://doi.org/10.3389/fmolb.2021.683132 Frontiers in Molecular Biosciences 8, pg 514, 2021 https://www.frontiersin.org/article/...lb.2021.683132. Creative Commons Attribution License (CC BY).
The nomenclature used to categorize chaperones is confusing. In general, chaperones, found in all cells, interact with unfolded or misfolded proteins and essentially catalyze their folding. They can do so by removing them from environments that would inhibit folding. Some can prevent aggregate formation or remove them. Some interact with proteins exiting the ribosome as they are being synthesized while some escort damaged proteins to sites of proteolysis.
Many chaperons are used to maintain proteostasis. They can be classified as to molecular size or groups based on overall structure and mechanism. We'll use the second approach. Here are some of the key player
Cage Chaperonins: Oliogomeric High Molecular Nanoparticles
Chaperonins, whose subunits can still be called chaperones, assemble into oligomeric, high molecular weight nanoparticle cages. These complexes are involved in the folding of larger proteins. The complexes consist of two stacked rings. Inside is an inner cavity in which larger proteins can fold in isolation (an easy way to remember the name chaperonin). A somewhat silly analogy would be a person dressing alone in a closet. These complexes are ATPase as the cleavage of ATP drives protein folding.
There are two main classes of chaperonins, Class I and Class II
Group I: These are found in bacteria, in some archaea and in mitochondria, which derived in evolution through an endosymbiotic relationship with bacteria. Proteins in this group include Hsp60, which forms a homo 14-mer, and the co-chaperone Hsp10, which forms a homo 7-mer. In E. Coli, hsp60 is also called GroEL, while the small Hsp10 is called GroES. GroEL and GroES together form the GroEL/GroES Complex, which is shown in the interactive iCn3D model in Figure \(\PageIndex{18}\) (note: it loads slowly given its large size).

Figure \(\PageIndex{18}\): GroEL/GroES Complex (1pcq). (Copyright; author via source).
Click the image for a popup (which loads slowly) or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?JNESMy2hS9ZBTST66
The 14 GroEL subunits form separate 7-mer (magenta) and 7-mer (gray) hollow rings, which stack on each other. The gray ring monomers each have an ADP/AlF3- (spacefill) bound. Seven GroES monomers, shown in cyan, form a "lid" over the end of the complex. This hetero 21-mer displays a C7 global symmetry. Lid binding requires ATP.
Figure \(\PageIndex{19}\) shows a mechanism for the GroEL/GroES protein folding cycle (shown in a linear arrangement).

In the left-most structure, a substrate protein (colored line drawing) binds in the cavity formed by the GroEL ring that does not have the GroES 7-mer cap (cyan). Rearrangement of the substrate protein and interactions with the interior wall trigger binding of ATP, binding of the top GroES cap on the cis end (same end as bound protein substrate), and release of the previously bound ADP, the distal trans GroES cap and a folded protein (not shown) from the trans end. Transient interactions and substrate protein conformation changes lead to the folding of the colored protein substrate within the time frame needed for the cleavage of the 7 bound ATPs. Binding of ATP and a new GroES cap at the bottom trigger release of the folded protein at the top along with the top GroES cap. The cycle then repeats with the binding of a new target protein substrate, ATP and a GroES cap leading to the dissociation of the ADP and the other GroES cap.
The sequestration of the bound protein substrate clearly prevents aggregation of the folding protein. The binding of any polymer into a confined small volume must be entropically disfavored since the polymer's conformational flexibility is reduced. Somehow an entropic activation energy barrier is reduced inside of the cage, maybe in a fashion similar to the restraining effects of disulfides on protein folding, which allows less conformational space to be explored on folding. GroEL has also been shown to bind in its hydrophobic cavity a fluorescent CdS semiconductor nanoparticle which can be released on addition and cleavage of ATP
Group 2: These are found in Archaea and in eukaryotes. The eukaryotic chaperonin in this group is CCT (cytosolic chaperonin containing TCP1) also known as TRic (tailless complex polypeptide 1 ring complex or TCP1-ring complex). These also contain two stacked rings but this time they are 8-mers of molecular weight 50-60K. This is similar to the Hsp60 monomers in the GroEL/GroES complex, but the monomer in Group 2 are not named with the beginning letters Hp. In addition, the complex does NOT have a cap like the GroES co-chaperone 7-mer in the GroEL/GroES complex. Rather, they have a built-in cap that closes on folding. The CCT/TRic chaperonin complex interacts with other "co-chaperones" including a prefoldin 6-mer complex, which inhibits aggregation and which "delivers" the protein substrate to CCT/TRic. It also binds to phosducin-like proteins. Well know substrate proteins for CCT/TRic include actin and tubulin (cytoskeletal proteins) and proteins involved in cell cycle control, but many other proteins (up to 10% of cytosolic proteins) might interact with it.
The genes encoding the 8 monomers in CCT/TRic arose from gene duplication and subsequent mutation so they are different from each other (technically they are called paralogs). The subunits are named α, β, ξ, δ, ε, ζ, η and θ. Figure \(\PageIndex{20}\) shows generalized structures of CCT/TRic.

Panel A shows a cryoEM of the complex with one subunit outlined in white and an adjacent PDB structure of the alpha subunit (1A6D).The red domain binds ATP. The relative arrangements of the monomers is shown in B.
The Human TRiC/CCT complex (7LUP) with a protein substrate, reovirus outer capsid protein sigma-3, bound in the internal cavity is shown in the interactive iCn3D model in Figure \(\PageIndex{21}\) (note: it loads slowly given its large size).

Figure \(\PageIndex{21}\): Human TRiC/CCT complex with reovirus outer capsid protein sigma-3 (7LUP) (Copyright; author via source).
Click the image for a popup (which loads slowly) or use this external link: https://structure.ncbi.nlm.nih.gov/i...Dxr3n6oy6zLsZ7
The cyan spacefill protein substrate is the encapsulated reovirus outer capsid protein sigma-3. The top ring 8-mers are shown in gray ribbon while the bottom ones are shown in different colors. It appears that each ring binds 4 ATPs for a total of 8 when the "attached lids" are closed.
Non-cage chaperones
A variety of chaperones that are not part of the nanoparticle chaperonin complex are also involved in protein folding in vivo. Some classify chaperones on the basis of molecular weight into 5 classes, Hsp60 (which we discussed above as monomers in the chaperonin cage complexes), Hsp70, Hsp90, Hsp104, and the small Hsps. We'll focus mostly in Hsp70 and its "co-chaperone", Hsp 40, and their bacterial analogs, DnaK and DnaJ, respectively.
Hsp70/Hsp 40: Hsp70 binds to hydrophobic regions of proteins which are more prevalent in unfolded and partially folded proteins and helps refold them through repeated cycles of binding and release, which is dependent on ATP cleavage. It also helps unfolded proteins through membranes and helps form/dissociate protein complexes. It's found in the cytoplasm and in a variety of organelles including chloroplasts, mitochondria, nuclei and the ER. There is a family of Hsp70 proteins (1, 1A, 1B, 2, 3, 4, 4L, 5–10, 12–18). Hsp70 proteins are made up of two regions. The nucleotide binding domain (NBD), which has intrinsic low ATPase activity, is located in the N-terminal region. The polypeptide substrate binding domain (SBD) is located in the carboxyl terminal end. It transiently recognizes short peptides in the target protein. A flexible stretch of conserved amino acids connects the two domains. The substrate binding domain has a beta sheet where peptide substrates bind and an alpha-helical region that acts like a lid.
From an evolutionary sense, they are one of the most conserved proteins. This shows the critical importance of protein folding to life. Some members of the Hsp70 family include DnaK (a bacterial Hsp70 that has been widely studied along with its Hsp40 co-chaperone partner, DnaJ) and BiP (HSPA5) that assists protein folding in the ER. and alpha crystalline in eukaryotes. Alpha crystalline comprises 30% of the lens proteins in the eye, where it functions, in part, to prevent nonspecific, irreversible aggregates. Some Hsp70 proteins are constitutively expressed, while others are induced by increased temperatures or other stress perturbants that affect protein structure, including radiation, inflammation and exposure to heavy metals. They are key to the maintenance of proteostasis.
Hsp70 proteins:
- bind to growing polypeptide chains as they are synthesized on ribosomes.
- express activity as monomers.
- have ATPase activity - i.e. they cleave the phosphoanhydride ATP (which can drive reactions).
- bind short, extended peptides, which stimulates the ATPase activity
- release bound peptides after ATP cleavage
Hsp70s are highly flexible so it is difficult to get detailed structural information comparing bound (to target peptides) and free states. Hsp70 appears to clamp onto hydrophobic regions of target proteins, making transient interactions characterized by multiple binding and release steps, until protein folding is complete. Hence the affinity for the Hsp70 proteins must be low enough (i.e. high dissociation constant KD where the KD is the inverse of the equilibrium binding constant as we will see in Chapter 5) for the target protein for many binding and release steps. The affinity of the SBD for the target protein is regulated by the nucleotide binding domain (NBD). When ADP is bound, the Hsp70 is in a closed state and has a high affinity for substrate, so the release rate for bound target protein is slow. When ATP is bound, Hsp 70 is in the open state in which the rate of association increases 100 fold and the rate of dissociation increases 1000 fold, so the affinity is lowered significantly. This enables multiple cycles of release, folding, and rebinding to occur.
As mentioned above, Hsp70s have intrinsic but low ATPase activity. When ATP is cleaved to ADP, Hsp70 returns to the closed, high-affinity state. Hence ATP hydrolysis affects both the binding of protein substrate) and kinetics of folding. This activity increases with the binding of protein substrates as well as with the binding of co-chaperones containing what is called a J domain (as is found in the bacterial co-chaperone DnaJ). Hence considerable conformational changing and signaling occur between the NBD and SBDs in a process we will call allosterism in Chapter xx.
The allosteric Hsp70 catalytic cycle is shown in Figure \(\PageIndex{22}\).

Figure \(\PageIndex{22}\): Hsp70 Catalytic Cycle for Protein Folding. Stetz G, Verkhivker GM (2015) Dancing through Life: Molecular Dynamics Simulations and Network-Centric Modeling of Allosteric Mechanisms in Hsp70 and Hsp110 Chaperone Proteins. PLoS ONE 10(11): e0143752. https://doi.org/10.1371/journal.pone.0143752. Creative Commons Attribution License,
Let's explore the catalytic cycle starting the left structure. (note that NBD subdomains are colored as follows: IA (in blue), IB (in red), IIA (in green), IIB (in cyan))
- Left structure (9 o'clock): ADP is bound so Hsp70 is in the closed (high affinity for protein substrate)state. The SBD consists of two parts or subdomains, the α-lid (magenta) section and the subdomain comprised of β-sheets (orange), where the actual protein substrate binds (not shown here). The α-helical lid of the SBD helps to keep the protein substrate from dissociating. The PDB ID for this structure is 2KHO, but if you explore this structure, no protein substrate is evident. The two domains of Hsp70, the SBD and the NBD, are connected by the flexible interdomain linker (black), are not interacting so they are shown in the domain undocked state.
- Top (12 o'clock): This represents an "intermediate" in which ATP is exchanging for bound ADP and the complex is changing to the open form, but without the release of protein substrate. Note that the α-lid is no longer interacting as tightly with the β-sheet subdomain of the SBD.
- Right (3 o'clock): The protein substrate (again not shown) is fully released, the α-lid is fully disengaged from the β-sheet subdomain of the SBD, and Hsp70 is in the open (to substrate binding) conformation. The SBD β-sheet subdomain and the NBD domains are still engaged and are shown in the domain-docked conformation.
- Bottom (6 o'clock): Weak protein substrate binding leads to an "intermediate" in which the α-lid is starting to engage with the β-sheet subdomain of the SBD. Substate binding promotes ATP hydrolysis, which occurs as Hsp70 returns to the full closed conformation (left structure, 9 o'clock) with bound ADP
Figure \(\PageIndex{23}\) shows the incredible conformational change that occurs on movement from the ADP-bound higher affinity closed form of E. Coli Hsp70 (DnaK) (2kho) to the ATP-bound lower affinity, domain-docked open(to substrate binding) form (4jne). Note how the alpha-helical lid in the left-hand side RBD moves to engage with the NBD on the right-hand side.
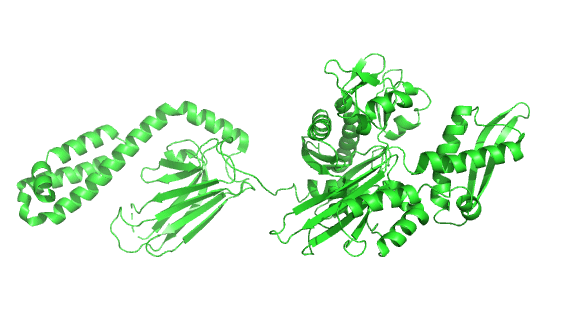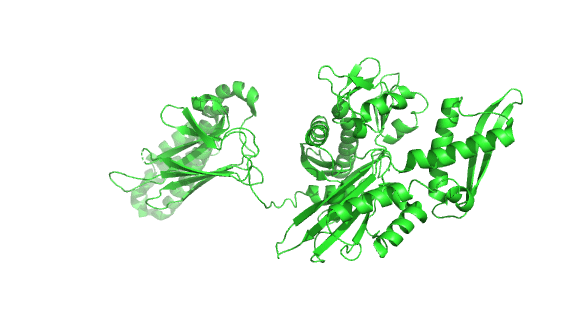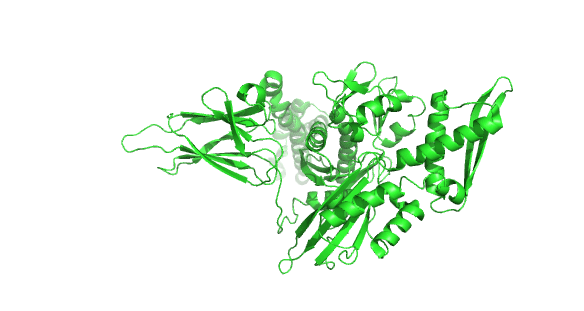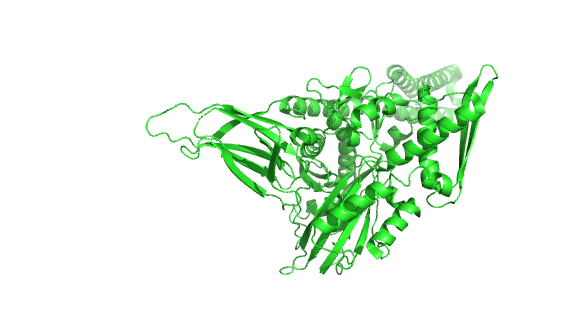
Figure \(\PageIndex{24}\) how the chaperonins and Hsp70/40 (DnaK/DnaJ) work in concert.

A few other chaperones acting downstream from Hsp70/Hsp40 and DnaK/DnaJ are shown in the image:
- TF (Trigger factor) and NAC/RAC: these are ribosome-binding chaperones
- Prefoldin (Pfd): bind unfolding proteins and shuffles them off to TRic.
- Hop: mediates interactions between Hsp70/Hsp40 and Hsp90
- Hsp90: This chaperone targets protein receptors (including steroid hormone receptors, protein kinases, and proteins with nucleotide-binding site and leucine-rich repeat (NLR) domains as are found in inflammasomes) and has a cycle similar to Hsp70 with some differences. Hsp90 has an N-terminal nucleotide binding domains, middle domains that interact with protein substrates, and C-terminal dimerization domains so the protein exists as a homodimer, When bound to ADP or in the absence of nucleotides, the homodimer forms an open V-shape conformation (as shown in the figure above). When ATP binds a huge conformation change occurs that closes the V shaped clamp. The protein has intrinsic ATPase activity, which reverses the conformational change. Many different co-chaperones bond to the HSP90 dimer and modulate the cycle at different points.
Additional Proteins also catalyze protein folding at key steps in the process. Here are two examples.
- Protein Disulfide Isomerase (PDI) - catalyzes the conversion of incorrect to correct disulfides. The active site consists of 2 sets of the the following sequence - Cys-Gly-His-Cys, in which the pKa of the cysteines are much lower (7.3) than normal (8.5). How would this facilitate disulfide isomerization?
- Peptidyl Prolyl-Isomerase - catalyzes X-Pro isomerization, by a mechanism, which probably involves bending the X-Pro peptide bond. How would this facilitate the process?
Many proteins have been found to possess PPI activity. One class is the immunophilins. These are small proteins found in the cytoplasm that bind anti-rejections drugs used to prevent tissue rejection after transplantation. The immunophilin FK506 binding protein (FKBP) binds FK506 while the protein cyclophilin binds that anti-rejection drug cyclosporin. The complex of cyclophilin:cyclosporin or FKBP:FK506 binds to an inhibits calcineurin, an important protein (with phosphatase activity) in immune cells (T cells) required for T cell function. In this case, immunophilin:drug binding to calcineurin inhibits the activity of the T cell, preventing immune attack on the transplanted tissue and rejection. The immunosuppressant drugs (FK506 and cyclosporin) inhibit the PPI activity of their respective immunophilin. The extent to which the PPI activity of cyclophiin is required for its activity is unclear, but it seems to be important for some of its biological effects.
Given the complexity of protein folding and the large number of chaperones involved in in vivo folding, it should not be a surprise that chaperone proteins bind to protein substrates through more than just hydrophobic interactions. Some chaperones (Spy and Trigger Factor) bind to charged regions on the surface of proteins, while the ER proteins calnexin and calreticulin bind to carbohydrates on glycoproteins. GroEL/GroES and TRiC/CCT also interact through electrostatic attractions with protein substrates. Nucleoplasm, a chaperone protein found in the nucleus, binds to histones which are strongly costively charged DNA binding proteins.
Unfolded Protein Response
As the site responsible for the folding of membrane proteins and proteins destined for secretion, as well as the major site for lipid synthesis, the endoplasmic reticulum (ER) must be able to maintain homeostatic conditions to ensure proper protein formation. Plasma cells that synthesize antibodies for secretion as part of the immune activation, show large increases in protein chaperones and ER membrane size
The main pathway controlling ER biology is the unfolded protein response (UPR) signaling pathway. If demand for protein synthesis in the ER exceeds capacity, unfolded proteins accumulate. This ER stress condition activates a protein called IRE1, a transmembrane Ser/Thr protein kinase (which phosphorylates proteins). IRE1 activates a transcription factor that controls the transcription of many genes associated with protein folding in the ER. Another protein, ERAD (ER-associated degradation) moves unfolded proteins back into the cytoplasm where they are degraded by the proteasome. Proteins involved in lipid synthesis are also activated as lipids are needed for membranes as the ER increases in size. If the stress can not be mitigated the signaling pathway leads to programmed cell death (apoptosis).
Schuck at al investigated the specific role and importance of UPR in the homeostasis of ER as modeled by the yeast Saccharomyces cervisiae. The UPR signaling pathway was analyzed using light and electron microscopy to visualize and quantify ER growth under various stress conditions. Western blotting procedures were performed to determine chaperone protein concentrations after stress induction and association with ER expansion after the ER was exposed to various treatment conditions. The authors found ER membrane expansion occurred through lipid synthesis since stress induction increased concentrations of proteins responsible for promoting lipid synthesis and expansion failed when the proteins were absent and lipid concentration was low. In addition, these lipid synthesis proteins were activated by the UPR signaling pathway. By separating ER size control and UPR signaling, they found that expansion occurred regardless of chaperone protein concentrations. However, if lipid synthesis genes were not available, raising the ER chaperone level helped alleviate stress levels in ER.
Redox Chemistry and Protein Folding
In general, we envision the interior of a cell to be in a reducing environment. Cells have sufficient concentrations of "b-mercaptoethanol"-like molecules (used to reduce disulfide bonds in proteins in vitro) such as glutathione (g-Glu-Cys-Gly) and reduced thioredoxin (with an active site Cys) to prevent disulfide bond formation in cytoplasmic proteins. For disulfide bonds to occur in a protein, a free sulfhydryl reacts with another one on a protein to form the more oxidized disulfide bond. This reaction occurs more readily if one of the Cys side chains had a lowered pKa (due to its immediate environment) making it a better nucleophile in the reaction. Most cytoplasmic proteins contain Cys with side chain pKa > 8, which would minimize disulfide bond formation as the Cys are predominantly protonated at that pH.
Disulfide bonds in proteins are typically found in extracellular proteins, where they serve to keep multisubunit proteins together as they become diluted in the extracellular milieu. These proteins destined for secretion are cotranslationally inserted into the endoplasmic reticulum (see below) which presents an oxidizing environment to the folding protein and where sugars are covalently attached to the folding protein and disulfide bonds are formed (see Chapter 3D: Glycoproteins - Biosynthesis and Function). Protein enzymes involved in disulfide bond formation contain free Cys which form mixed disulfides with their target substrate proteins. The enzymes (thiol-disulfide oxidoreductases, protein disulfide isomerases) have a Cys-XY-Cys motif and can promote disulfide bond formation or their reduction to free sulfhydryls. They are especially redox-sensitive since their Cys side chains must cycle between and free disulfide forms.
Intracellular disulfide bonds are found in proteins in the periplasm of prokaryotes and in the endoplasmic reticulum (ER) and mitochondrial intermembrane space (IMS) of eukaryotes. For these proteins, the beginning stage of protein synthesis (in the cytoplasm) is separated temporally and spatially from the site of disulfide bond formation and final folding. Disulfide bonds can be generated in a target protein by concomitant reduction of a disulfide in a protein catalyst, leaving the net number of disulfides constant (unless the enzyme is reoxidized by an independent process). Alternatively, a disulfide can be formed by the transfer of electrons to oxidizing agents such as dioxygen.
In the ER, disulfide bond formation is catalyzed by proteins in the disulfide isomerase family (PDI). To function as catalysts in this process, the PDIs must be in an oxidized state capable of accepting electrons from the protein target for disulfide bond formation. A flavoprotein, Ero1, recycles PDI back to an oxidized state, and the reduced Ero1 is regenerated by passing electrons to dioxygen to form hydrogen peroxide. In summary, on formation of disulfides in the ER, electrons flow from the nascent protein to PDIs to the flavin protein Ero1 to dioxgen (i.e. to better and better electron acceptors). The first step is really a disulfide shuffle, which, when coupled to subsequent steps, leads to de novo disulfide bond formation.
In the mitochondria, disulfide bond formation occurs in the intermembrane space (IMS) and is guided by the mitochondria disulfide relay system. This system requires two important proteins: Mia40 and Erv1. Mia40 contains a redox-active disulfide bond cys-pro-cys and oxidizes cys residues in polypeptide chains. Erv1 can then reoxidize Mia40 which can in turn get reoxized by the heme in cytochrome c. Reduced cytochrome C is oxidized by cytochrome C oxidase of electron transport through the transfer of electrons to dioxygen to form water. The importance of IMS protein oxidation is less understood, but it is believed that the oxidative stress caused by a dysfunction could lead to neurodegenerative diseases.
A recent review by Riemer et al compares the ER and mitochondrial processes for disulfide bond formation:
- Many more and diverse proteins form disulfides in the ER compared to the IMS. Most in the IMS have low molecular mass and have two disulfide bonds between helix-turn-helix motifs. These protein substrates include chaperones that facilitate the localization of proteins in the inner membrane, and in proteins involved in electron transport in the inner membrane.
- There are many PDIs in the ER, probably reflecting the structural diversity of protein substrates in the ER. However, Mia40 appears to be the only PDI in the IMS.
- "De novo" disulfide bond formation is initiated by Ero1 in the ER and Erv1 in the IMS. Convergent evolution led to similar structures for both - a 4-helix bundle that binds FAD with two proximal Cys.
- The mitochondria pathway leads to water formation on reduction of dioxygen, not hydrogen peroxide, minimizing the formation of reactive oxygen species in the mitochondria. The peroxide formed in the ER is presumably converted to an inert form.
- The IMS is in more intimate contact with the cytoplasm through outer membrane proteins called porins which would allow some glutathione access. The IMS presents a more oxidizing environment than the cytoplasm (with more glutathione). The ER, without a porin analog, would be more oxidizing.
- The reversible formation of disulfides in the ER regulates protein activity.
Disulfide bond regulation in the Periplasmic Space of Bacteria
The redox sensitivity of the Cys side chain found in disulfide bonds is important in regulating protein activity. In particular, the thiol group of the amino acid Cys, an important nucleophile often found in the active site, can be modified to control protein activity. The formation of a disulfide bond or the oxidation of free thiols to sulfenic acid or further to sulfinic or sulfonic acid can block protein activity. The E. Coli periplasmic protein DsbA (disulfide bond A) converts adjacent free thiols into disulfide-linked Cystine, in the process becoming reduced. DsbB is reoxidized by DsbA back to its catalytically active form. What about periplasmic protein like YbiS with an active site Cys? Since the environment of the periplasm is oxidizing, YbiS is protected from oxidative conversion of the free Cys to either sulfinic or sulfonic acids causing the protein to become inactive. The mechanism involves two periplasmic proteins known as DsbG and DsbC which are similar to thioredoxin. These two proteins are able to donate electrons to the unprotected thiol preventing it from becoming oxidized, which allows YbiS to remain active in the periplasm. To maintain activity, DsbG and DsbC are reduced by another periplasmic protein, DsbD.
Protein Transport Across Membranes
How does a protein "decide" its final location after synthesis? Protein synthesis occurs in the cytoplasm, but proteins may end up outside of the cell, in cell membranes, internalized into various organelles, or remain in the cytoplasm. How is the decision made? There must be signals in the protein which target proteins to various sites in a cell, where processing can occur. Proteins that are destined for secretion or plasma membrane insertion typically have a signal peptide at the N-terminus which binds to a signal recognition particle in a cotranslational process, which temporarily arrests translation and nascent folding. This complex docks to signal recognition complex docking sites in the endoplasmic reticulum membrane, where translation continues as the nascent polypeptide extends through a protein pore in the ER membrane. Transport across the ER membrane can also occur partially or fully in a post-translational process if nascent proteins are partially or fully folded through interactions with cytoplasmic chaperones such as Hsp70/40. Figure \(\PageIndex{25}\) shows the cotranslational (a) and post-translation (b) pathways for uptake into the ER lumen.

In both (a) and (b), the protein is shown during synthesis as it is bound to the ribosome (40S/60S) nanoparticles. The cytoplasmic signal recognition particle (SRP) binds to the hydrophobic signal sequence of the nascent protein. The signal is typically at or near the N-terminus of the growing protein. Either co- or post-translationally, the nascent protein is delivered to Sec61, which is both a ribosome receptor and a gated pore for passages of the target protein. Sec61 is part of a larger ER translocon complex which also includes Sec62 and Sec63. The membrane topology and subunit structure of the Sec proteins are shown in (c). Addition proteins including the chaperones BiP Hsp70/40 are also shown.
Protein transport across the endoplasmic reticulum membrane. Mechanism of (a) co-translational and (b) posttranslational transport of precursor proteins through the Sec61 channel. (c) Topological domains of Sec61α1/ß/γ, (d) Sec62 and (e) Sec63. We note that (i) Sec63 interacts with Sec62 involving a cluster of negatively charged amino-acid residues near the C terminus of Sec63 and positively charged cluster in the N-terminal domain of Sec62, (ii) Sec62 interacts with the N-terminal domain of Sec61α via its C-terminal domain, (iii) BiP can bind to ER luminal loop 7 of Sec61 α via its substrate-binding domain and mediated by the ATPase domain of BiP and the J-domain in the ER luminal loop of Sec63, (iv) Ca2+-CaM can bind to an IQ motif in the N-terminal domain of Sec61α and (v) LC3 can bind to a LIR motif in the C-terminal domain of Sec62. 40S, 40S ribosome subunit; 60S, 60S ribosome subunit; SR, heterodimeric SRP receptor; SRP, signal recognition particle.
Figure \(\PageIndex{26}\) shows an interactive iCn3D model of the Sec Complex from yeast (6ND1).
.png?revision=1)
Figure \(\PageIndex{26}\): Sec Complex from yeast (6ND1). (Copyright; author via source).
Click the image for a popup (which loads slowly) or use this external link: https://structure.ncbi.nlm.nih.gov/i...6VoV8gTiUCQko8
The model shows Sec 61 and SEc 63 from Figure 25 above. Sec 61 is the ER protein-conducting channel (the analog in prokaryotes is Sec Y).
The model is colored as follows:
- Protein transport protein SEC61 (B)- magenta
- Protein translocation protein SEC63 (A) - blue
- Protein transport protein SSS1 - darker brown
- Protein transport protein SBH1 - green
- Translocation protein SEC66 - gold
- Translocation protein SEC72 - salmon
If the signal sequence of the protein to be imported not very hydrophobic, it doesn't bind the signal recognition particle, and hence is imported post-translationally. For these proteins, the Sec61 channel requires additional proteins, Sec62 and Sec63. These required also the BiP Hsp70/40 ATPase for import. Sec63 opens a gate on Sec61 leading to a wide opening, allowing proteins into the lipid bilayer.
If destined for secretion, a protein enters the lumen of the ER. Proteins destined for insertion into the cell surface membrane gets "stuck" in the ER membrane, and through a process of vesiculation merges with the Golgi and eventually with the cell surface membrane. Proteins that are taken into organelles like mitochondria are done so in a post-translational process that requires facilitation by protein chaperones. Final protein folding occurs inside the organelle. In both cases, nonnative proteins pass through the membrane after which final folding occurs.
An intriguing question is how the decision is made to keep a protein either in the membrane or allow it to pass through completely (in the case of proteins destined for secretion). Hessa et al investigated this "decision-making" process by studying the eukaryotic membrane pore protein complex, Sec 61 translocon (show in the above figures), whose activity must be closely regulated with the folding of the growing protein. In studying this process, they considered three local regions in a membrane: the hydrophobic region comprised of the nonpolar acyl tails of membrane lipids, the interfacial region in the vicinity of the polar head groups, and the aqueous regions (bulk water) on each side of the head groups. A 19 amino acid peptide was used as the experimental model protein which was added to the translocon. This size was chosen since it is just long enough to span the hydrophobic part of the membrane if the peptide were in an alpha-helical conformation (which is common in membrane-spanning proteins). They varied the proportion of amino acids that tend to partition into each of three regions and studied the disposition of the peptide after interaction with membrane and translocon. To test if the results were consistent with the thermodynamics of amino acid partitioning into nonpolar environments (and not kinetic considerations), they used the Wimley and White hydrophobicity scale, based on the free energy of transfer of amino acid side chains into nonpolar environments, to predict target peptide disposition with the membrane. The table below shows the propensity of amino acids to be in each region at equilibrium, based on this hydrophobicity scale.
Table: Amino Acid Partitioning Into Membrane Regions
| Region | Amino Acids |
| Bulk water | Arg, Asn, Asp, Gln, Glu, His, Lys, Pro |
| Bulk water + interfacial | Ala, Cys, Gly, Ser, Thr |
| Interfacial | Tyr |
| Hydrophobic | Ile, Leu, Met, Phe, Trp, Val |
Their experimental results were in concordance with those predicted using the above scale. If a polyalanine 19 mer was used, no insertion was observed. With five leucines in the peptide, almost 90% was inserted into the membrane. The results would be modeled using a two-state equilibrium: Peptide inserted ↔ Peptide translocated.
They then substituted each of the twenty amino acids into a given position into a target peptide and used the results to develop an empirical scale for membrane transfer, not one based on the simple transfer to nonpolar medium. This new scale matched the hydophobicity scale, suggesting insertion and transfer decisions were based on the thermodynamics of side chain partitioning. They also varied the position of the varied amino acid in the test peptide. If the amino acid favored the bulk and/or interfacial region, the peptide would be inserted if that amino acid were at the end of the peptide, not the middle. For translocation, the peptide had to be amphiphilic with one face polar and the other nonpolar.
They developed a simple equilibrium model to show the processes involved, as shown below in a top-down view of the membrane in Figure \(\PageIndex{27}\).
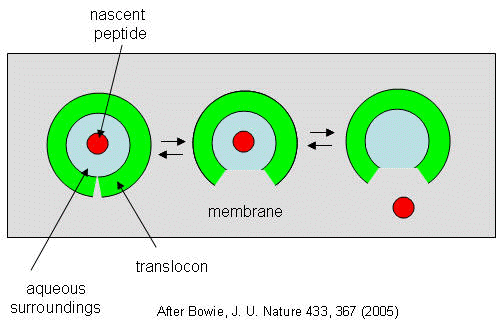
The translocon, shown in green, has a water-filled pore but also a sidewise opening toward the membrane interior. The target peptide enters the pore. Transient conformational changes in the pore expose the peptide to the nonpolar membrane core. The target peptide samples both the aqueous and nonpolar environments and partitions into them based on the considerations mentioned above. If it partitions more favorably into the hydrophobic core, it will do so and cause the peptide to become membrane-bound. Otherwise, it will pass through to the other side. This can be modeled as an equilibrium process if the rate of translocation is slow compared to the rates of translocon conformational change and environmental sampling by the peptide. Obviously, the process becomes more complicated if the target is a large protein.
Bacterial toxin proteins also have evolved ways to pass through a cell membrane, again in a nonnative state, through a protein channel in the membrane. Krantz et al have recently worked out details of how the anthrax toxin protein moves through eukaryotic cell membranes. Three anthrax proteins are involved. One is a "prepore" protein that binds to specific proteins on the cell membrane, where it is activated by limited proteolysis to form a pore protein, which assembles into the homoheptamer prepore in the membrane. Two other proteins secreted by the bacteria, lethal factor and edema factor, bind to the heptamer complex and the whole assembly is then taken up into the cell by invagination to form a vesicle with the pore complex in the membrane. This vesicle fuses with a lysosome in the cell, and upon acidification, a conformational change occurs in the prepore complex to activate it. The lethal and edema factors unfold partially, possibly to a molten globule state, and are then passed through the pore into the cell where they exert their toxic influences. An electrochemical potential gradient (which we will discuss later in the semester) is required for the passage of the factors through the membrane. The active pore further unravels the factor protein, facilitating transport.
Krantz et al. studied the pore protein by mutating two amino acids, Phe427 and Ser 429, on each monomer of the pore to Cys. They then modified the Cys with [2-(trimethylammonium) ethylmethanethiosulfonate and observed effects on ion conductance of the pore and pore conformations. They noted that when both residues were mutated and chemically modified, that ion conductance was blocked, suggesting that these side chains were localized in the narrowest part of the channel. When Phe 427 alone was mutated to smaller side chains (Ala), ion conductance increased but the transfer of peptides from the factor proteins was inhibited. This suggested that an aromatic ring in the narrow part of the channel opening participates in the translocation of bacterial proteins through the membrane. They then analyzed the transport of a variety of small molecules with varying hydrophobicity through the wild-type pore. Their results were consistent with the binding of the molecules through hydrophobic and aromatic electron interactions. They suggest a mechanism of transport consistent with their data in which the unfolded protein "ratchets" through the pore, which promotes factor protein unfolding to expose more hydrophobic groups to the nonpolar aromatic ring in the pore. This mechanism is similar to how the chaperone complex GroEL/GroES unfolds protein in its large central cavity in a process which requires hydrolysis of ATP, not a transmembrane potential. In addition, the Sec61 translocon in the inner membrane of bacteria and in eukaryotic ER membranes also has a pore containing a ring of hydrophobic groups (Ile).