
29.2: Regulation of glucose metabolism from a liver-centric perspective
- Page ID
- 53877


Han, HS., Kang, G., Kim, J. et al. Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med 48, e218 (2016). https://doi.org/10.1038/emm.2015.122
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Experimental & Molecular Medicine volume 48, pagee218 (2016)
Abstract
Glucose homeostasis is tightly regulated to meet the energy requirements of the vital organs and maintain an individual’s health. The liver has a major role in the control of glucose homeostasis by controlling various pathways of glucose metabolism, including glycogenesis, glycogenolysis, glycolysis and gluconeogenesis. Both the acute and chronic regulation of the enzymes involved in the pathways are required for the proper functioning of these complex interwoven systems. Allosteric control by various metabolic intermediates, as well as post-translational modifications of these metabolic enzymes constitute the acute control of these pathways, and the controlled expression of the genes encoding these enzymes is critical in mediating the longer-term regulation of these metabolic pathways. Notably, several key transcription factors are shown to be involved in the control of glucose metabolism including glycolysis and gluconeogenesis in the liver. In this review, we would like to illustrate the current understanding of glucose metabolism, with an emphasis on the transcription factors and their regulators that are involved in the chronic control of glucose homeostasis.
Overview of glucose metabolism in the liver
Under feeding conditions, dietary carbohydrates are digested and processed by various glucosidases in the digestive tract, and the resultant monosaccharides, mainly hexose glucose, are transported into various tissues as a primary fuel for ATP generation.1 In most mammalian tissues, the catabolism of glucose into pyruvate, termed glycolysis, is preserved as a major pathway in eliciting ATP. In tissues with abundant mitochondria, cytosolic pyruvate is transported into the mitochondrial matrix, converted to acetyl-CoA by pyruvate dehydrogenase complex, and incorporated into the tricarboxylic acid cycle in conjunction with oxaloacetate. The cycle generates energy equivalent to ATP (that is, GTP) as well as both NADH and FADH2, which serve as important electron carriers for electron transport chain-oxidative phosphorylation, resulting in the generation of ATP.
In some cases, such as red blood cells lacking mitochondria or cells under ischemic conditions, pyruvate is converted into lactate in the cytosol to regenerate the NAD+ that is necessary for the continued generation of ATP by substrate-level phosphorylation via anaerobic glycolysis. Excessive carbohydrates in the liver are first converted into glycogen, a storage form of glucose in animals, by glycogenesis. In addition, in a carbohydrate-rich diet, the excessive carbohydrates are also converted into fatty acids via lipogenesis using the acetyl-CoA generated from glycolysis-driven pyruvate, which is incorporated into very low density lipoproteins for transport to white adipose tissue for the storage.2 The regulation of glycogen metabolism is examined in detail in this section, and the transcriptional control of glycolysis and lipogenesis is delineated in the following section.
Under fasting conditions, the liver has a major role in generating glucose as a fuel for other tissues, such as the brain, red blood cells and muscles. Initially, an increase in the pancreatic hormone glucagon initiates the cascade of kinase action (stated below in detail) that releases glucose from the stored glycogen via glycogenolysis.1 Normally, stored glycogen is critical for maintaining glucose homeostasis in mammals during an overnight fasting period. During a longer term fast or starvation, essentially all of the stored glycogen in the liver is depleted (after ~30 h of fasting), and de novo glucose synthesis or gluconeogenesis is responsible for the generation of glucose as a fuel for other tissues. Major non-carbohydrate precursors for gluconeogenesis are lactate, which is transported from peripheral tissues such as skeletal muscles or red blood cells, and glycerol, which is released from the adipose tissues via enhanced lipolysis during fasting. Most of the initial precursors for gluconeogenesis are generated in the mitochondria (except glycerol 3-phosphate via glycerol kinase activity), but the majority of the reaction occurs in the cytosolic part of the cell. The complex regulatory mechanism is delineated in detail in the following section, with an emphasis on the transcriptional control of key regulatory enzyme genes.
Regulation of glycogen metabolism in the liver
The accumulation of glycogen in the liver during feeding conditions provides a storage form of glucose that can be used in times of reduced food intake (Figure 1). Multiple layers of regulation are required for this process for both the activation of glycogen synthase, which is a key enzyme of glycogenesis (glycogen synthesis), and the inhibition of glycogen phosphorylase, which is a key enzyme of glycogenolysis (glycogen breakdown) in the liver. Glycogen synthase is a major enzyme that facilitates the elongation of glycogen chains by catalyzing the transfer of the glucose residue of UDP-glucose to the non-reducing end of a pre-existing glycogen branch to produce a new α1→4 glycosidic linkage. The regulation of glycogen synthase has been mostly studied using a muscle-specific isoform. In the muscle, glycogen synthase is inactivated via phosphorylation on multiple serine residues by various serine/threonine kinases such as casein kinase-1, protein kinase A (PKA), and glycogen synthase kinase-3 (GSK-3). Most notably, the phosphorylation of glycogen synthase by GSK-3 at serine residues 640, 644 and 648 has been linked to the most important inhibitory post-translational modification for its catalytic activity.
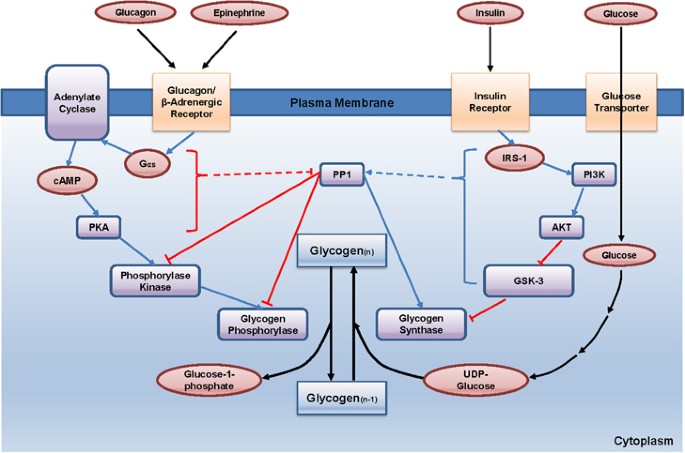
Regulation of hepatic glycogen metabolism. Under fasting conditions, glucagon and epinephrine induce cAMP-dependent signaling cascades, leading to the activation of glycogen phosphorylase and glycogenolysis while inhibiting glycogenesis. Conversely, feeding enhances insulin-mediated signaling in the liver, leading to the activation of both PP1 and Akt, thus promoting glycogen synthesis in response to increased glucose uptake in the liver. See the main text for more specific regulatory pathways. cAMP, cyclic AMP.
Under fasting conditions, dephosphorylated and active GSK-3 phosphorylate and inactivate glycogen synthase, leading to the inhibition of hepatic glycogen synthesis. On feeding, increased insulin signaling activates Akt in the cell, which in turn phosphorylates and inactivates GSK-3, thus resulting in the activation of glycogen synthase. In addition, increased concentrations of glucose 6-phosphate allosterically activate this enzyme, thus potentiating its catalytic activity under feeding conditions.3, 4 One recent publication argues against the role of GSK-3 in the regulation of the liver-specific isoform of glycogen synthase. In that study, Guinovart et al.5 mutated the corresponding serine residues that are shown to be regulated by GSK-3 in the liver-isoform of glycogen synthase. They found that the mutation of those residues did not affect the overall enzyme activity but that the mutation of serine 7 to alanine, a site that is recognized and regulated by PKA, led to the increased activity of this enzyme. Further study is necessary to determine whether these results can be verified in vivo using animal models such as liver-specific knock-in mice for S7A liver glycogen synthase. The protein phosphatase 1 (PP1) may be responsible for the dephosphorylation and activation of glycogen synthase. Accordingly, both glucose and insulin have been shown to activate PP1 activity, whereas glucagon and epinephrine have been linked to the inhibition of its activity.
Glycogen phosphorylase is a major enzyme involved in glycogenolysis (Figure 1). This enzyme catalyzes the reaction of the removal of a glucose residue from the non-reducing end of a glycogen chain, leading to the generation of glucose 1-phosphate.6 Glucose 1-phosphate can be converted into glucose 6-phosphate by phosphoglucomutase, and glucose 6-phosphate can be incorporated into glycolysis or further converted into glucose by glucose 6-phosphatase, depending on the energy status of the organism. Glycogen phosphorylase is active when it is phosphorylated at its serine 14 residue. The phosphorylation of glycogen phosphorylase requires a cascade mechanism of epinephrine and glucagon in the liver. On the activation of Gαs by the binding of hormones to cell surface G protein-coupled receptors (beta adrenergic receptor or glucagon receptor), the intracellular cyclic AMP (cAMP) levels increase via adenylate cyclase, leading to the activation of PKA. PKA is then responsible for the phosphorylation and activation of glycogen phosphorylase kinase, which in turn phosphorylates and activates glycogen phosphorylase to enhance glycogen breakdown. Under feeding conditions, this kinase cascade is inactive due to the lack of secretion of catabolic hormones. In addition, insulin promotes the activation of PP1, which dephosphorylates and inactivates glycogen phosphorylase. In essence, the anabolic hormone insulin promotes glycogenesis and inhibits glycogenolysis via the activation of PP1, leading to the dephosphorylation of glycogen phosphorylase (inactivation) and glycogen synthase (activation), and via the activation of Akt, leading to the phosphorylation of GSK-3 (inactivation) that is unable to phosphorylate and inactivate glycogen synthase.
Control of hepatic glycolysis
As stated above, glycolysis is critical to the catabolism of glucose in most cells to generate energy. The key rate-limiting enzymes for this pathway include glucokinase (GK, also termed hexokinase IV), which converts glucose into glucose 6-phosphate; phosphofructokinase-1 (PFK-1), which converts fructose 6-bisphosphate into fructose 1,6-bisphosphate; and liver-type pyruvate kinase (L-PK), which converts phosphoenolpyruvate (PEP) into pyruvate in the liver. These enzymes are tightly regulated by allosteric mediators that generally promote the catabolism of glucose in the cell.2, 7, 8, 9
GK is a high Km hexokinase that is present in the liver and the pancreatic beta cells, thus functioning as a glucose sensor for each cell type. Unlike the other hexokinase isotypes, GK activity is not allosterically inhibited by its catalytic product, glucose 6-phosphate in the cell, thus enabling the liver to continuously utilize glucose for glycolysis during conditions of increased glucose availability, such as during feeding conditions. GK is regulated via its interaction with glucokinase regulatory protein (GKRP). In the low intracellular glucose concentration during fasting, the binding of GK and GKRP is enhanced by fructose 6-phosphate, leading to the nuclear localization of this protein complex. Higher concentrations of glucose during feeding compete with fructose 6-phosphate to bind this complex, which promotes the cytosolic localization of GK that is released from GKRP, thus causing the increased production of glucose 6-phosphate in this setting.10
PFK-1 catalyzes the metabolically irreversible step that essentially commits glucose to glycolysis. This enzyme activity is allosterically inhibited by ATP and citrate, which generally indicate a sign of energy abundance. Reciprocally, it is allosterically activated by ADP or AMP, making it more efficient to bring about glycolysis to produce more ATP in the cell. In addition, PFK-1 activity is allosterically activated by fructose 2,6-bisphosphate (F26BP), a non-glycolytic metabolite that is critical for the regulation of glucose metabolism in the liver. F26BP is generated from fructose 6-phosphate by the kinase portion of a bifunctional enzyme that contains both a kinase domain (phosphofructokinase-2, PFK-2) and a phosphatase domain (fructose 2,6-bisphosphatase, F-2,6-Pase). PFK-2 is activated by the insulin-dependent dephosphorylation of a bifunctional enzyme that activates PFK-2 activity and simultaneously inhibits F-2,6-Pase activity to promote the increased F26BP concentration. Glucagon-mediated activation of PKA is shown to be responsible for the phosphorylation and inactivation of the kinase portion of this enzyme.7
Unlike its muscle counterpart, L-PK is also a critical regulatory step in the control of glycolysis in the liver. As in the case of other glycolytic enzymes, L-PK activity is regulated by both allosteric mediators and post-translational modifications. L-PK activity is allosterically activated by fructose 1,6-bisphosphate, an indicator for the active glycolysis. By contrast, its activity is allosterically inhibited by ATP, acetyl-CoA, and long-chain fatty acids, all of which signal an abundant energy supply. Additionally, the amino acid alanine inhibits its activity, as it can be readily converted to pyruvate by a transamination reaction. L-PK is inhibited by PKA following a glucagon-mediated increase in intracellular cAMP during fasting and is activated by insulin-mediated dephosphorylation under feeding conditions.7
In addition to the acute regulation of key regulatory enzymes, glycolysis is regulated by a transcriptional mechanism that is activated during feeding conditions. Two major transcription factors, sterol regulatory element binding protein 1c (SREBP-1c) and carbohydrate response element binding protein (ChREBP), are responsible for the transcriptional activation of not only glycolytic enzyme genes but also the genes involved in fatty acid biosynthesis (such as fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and stearoyl-CoA desaturase 1 (SCD1)) and triacylglycerol formation (such as glycerol 3-phosphate acyltransferase (GPAT) and diacylglycerol acyltransferase 2 (DGAT2)), a process that is normally activated by a carbohydrate-rich diet (Figure 2).11 Because these processes are often coordinately regulated, that is activated during feeding and inhibited by fasting, they are sometimes collectively called lipogenesis.
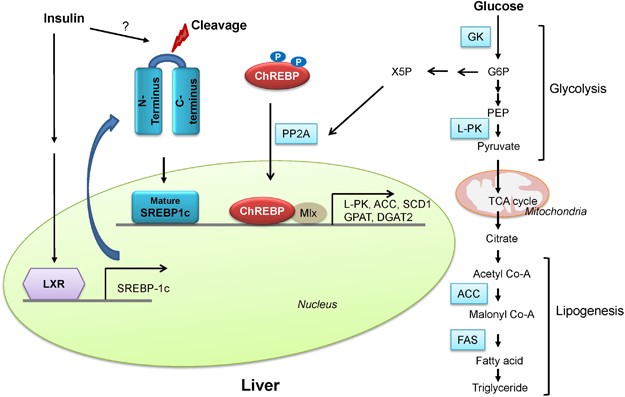
Regulation of hepatic glycolysis. Under feeding conditions, increased glucose uptake in hepatocytes promotes glycolysis and lipogenesis to generate triglycerides as storage forms of fuel. This process is transcriptionally regulated by two major transcription factors in the liver, SREBP-1c and ChREBP-Mlx heterodimer, which mediate the insulin and glucose response, respectively. See the main text for more specific regulatory pathways.
SREBPs are the major regulators of lipid metabolism in mammals. They are members of the basic helix-loop-helix leucine zipper (b/HLH/LZ) type transcription factor families comprising SREBP-1a, SREBP-1c, and SREBP-2. SREBP is translated as an endoplasmic reticulum (ER)-bound precursor form that contains the N-terminal transcription factor domain and the C-terminal regulatory domain linked with the central transmembrane domain.12 Within this family of transcription factors, SREBP-2 is linked to the control of cholesterol uptake or biosynthesis in the liver by the transcriptional activation of the genes involved in the pathway including low density lipoprotein receptor (LDLR), 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), hydroxy-3-methylglutaryl-coenzyme A synthase 1 (HMGCS1), and farnesyl diphosphate synthase (FDPS). SREBP-1c, however, activates the genes encoding the enzymes for lipogenesis (FAS, ACC, SCD1, and DGAT2) as well as GK, which is a first enzyme in the commitment step of glucose utilization in the liver. Indeed, liver-specific SREBP-1c knockout mice showed an impaired activation of lipogenic genes in a high carbohydrate diet, thus confirming the importance of this transcription factor in the regulation of hepatic glycolysis and fatty acid biosynthesis.13 SREBP-1a is not highly expressed in the liver but was shown to be involved in the formation of inflammasomes in response to lipopolysaccharide (LPS) treatment in macrophages by transcriptional activation of Nlrp1.14 The regulation of SREBP-2 and SREBP-1c are quite distinct in the liver. The expression of SREBP-2 is not controlled by sterols, but its proteolytic processing is tightly regulated by intracellular concentrations of cholesterol. It is normally bound in the ER via the interaction of SREBP-cleavage-activating protein (SCAP) and insulin-induced gene protein (INSIG) in the presence of high intracellular cholesterol levels, and the reduction in the cholesterol concentration releases the interaction of SCAP and SREBP-2/INSIG complex, resulting in the translocation of the latter complex into the Golgi apparatus and the liberation of the active SREBP-2 factor by sequential proteolytic cleavages.15 Unlike SREBP-2, SREBP-1c is mainly regulated at the transcription level by insulin. The exact transcription factor that mediates this insulin-dependent signal is not yet clear, although SREBP-1c itself might be involved in the process as part of an auto-regulatory loop. Interestingly, the oxysterol-sensing transcription factor liver X receptor (LXR) is shown to control the transcription of SREBP-1c, suggesting that SREBP-1c and SREBP-2 could be regulated differently in response to cellular cholesterol levels.16 Recent studies have revealed the involvement of various kinases in the control of SREBP-1c activity. In HepG2 cells, PKA was shown to reduce the DNA binding ability of SREBP-1a by the phosphorylation of serine 338 (equivalent of serine 265 for SREBP-1c).17 A report by Bengoechea and Ericsson suggested that GSK-3, a kinase known to reduce glycogen synthesis by targeting glycogen synthase, downregulates SREBP-1 activity via the phosphorylation of the C-terminal residue that promotes the ubiquitin ligase Fbw7-dependent degradation of SREBP-1 proteins.18 In addition, both AMP activated protein kinase (AMPK) and its related kinase salt-inducible kinase (SIK) 1 are involved in the down-regulation of its activity through inhibitory phosphorylation (serine 372 for AMPK, which blocks proteolysis and nuclear localization of SREBP-1c, and serine 329 for SIK1, which directly reduces its transcriptional activity).19, 20 These data suggest that the fine-tuning of SREBP-1c activity is critical to the maintenance of glucose and lipid homeostasis in the liver.
The other prominent transcription factor for controlling glycolysis and fatty acid biosynthesis in the liver is ChREBP. ChREBP was initially known as Williams-Beuren syndrome critical region 14 (WBSCR14) and was considered one of the potential genes that instigate Williams-Beuren syndrome. Later, by using a carbohydrate response element (ChoRE) from L-PK, ChREBP was isolated as a bona fide transcription factor for binding ChoRE of glycolytic promoters.21 Indeed, ChREBP is highly expressed in tissues that are active in lipogenesis such as the liver, brown adipocytes, white adipocytes, small intestine, and kidney. As in the case for SREBP, ChREBP belongs to the b/HLH/LZ transcription factor family and forms a heterodimer with another b/HLH/LZ transcription factor Max-like protein X (Mlx) on the glycolytic promoter.22 As in the case for the SREBP-1c, the expression of ChREBP is increased in the liver as a result of a high carbohydrate diet, and the effect was recapitulated in primary hepatocytes with high glucose treatment.
A recent report indeed suggested a role for LXR in the transcriptional activation of ChREBP in response to glucose, although the study needs to be further verified because the transcriptional response is shown not only by the treatment of D-glucose, a natural form of glucose present in animals, but also by the treatment of unnatural L-glucose, a form of glucose that is not known to activate lipogenesis in the liver.23 Moreover, studies performed in LXR knockout mice revealed no changes in ChREBP expression in the liver, arguing against the role of LXR in the control of ChREBP.24 Glucose is also shown to regulate ChREBP activity by controlling its nuclear localization. There are three prominent serine/threonine residues that are targeted by serine/threonine kinases. PKA is shown to phosphorylate serine 196, which is critical for cellular localization, and threonine 666, which is critical for its DNA binding ability, whereas AMPK phosphorylate serine 568 dictates its DNA binding ability. All three sites are phosphorylated under fasting conditions by these kinases and are dephosphorylated under feeding by xylulose 5-phosphate (X5P)-mediated activity of protein phosphatase 2A (PP2A).25, 26 However, the current model needs to be further verified due to the contrasting data that have been published regarding the role of these phosphorylations on ChREBP activity.
First, high glucose concentrations in primary hepatocytes do not result in decreased cAMP levels or PKA activity, suggesting that other signals might be necessary to mediate the high glucose-dependent nuclear translocation of ChREBP. In addition, a serine to alanine mutant of ChREBP still requires high glucose for its full activity, suggesting that additional actions are necessary to recapitulate the high glucose-mediated activation/nuclear localization of ChREBP in the liver.27, 28 The physiological role of ChREBP in liver glucose metabolism was verified by in vivo studies. ChREBP knockout mice were born in a Mendelian ratio and showed no developmental problems. The knockout animals showed reduced liver triacylglycerol levels together with a reduction in lipogenic gene expression, thus confirming the role of ChREBP in the control of hepatic glycolysis and fatty acid synthesis.29 Interestingly, the compensatory increase in glycogen was observed in the livers of these mice, suggesting that these mice adapted to store more glycogen as a storage form of fuel as opposed to triacylglycerol. In ob/ob mouse liver, increased accumulation of nuclear ChREBP was shown, suggesting that this phenomenon might be causal to the fatty liver phenotype in these mice. Indeed, knockdown of ChREBP in ob/ob mice reduced the rate of lipogenesis with decreased expression of most lipogenic genes.30 Furthermore, the depletion of hepatic ChREBP in ob/ob mice improved hyperglycemia, hyperlipidemia, and hyperinsulinemia, suggesting that regulation of ChREBP might be critical in the control of metabolic disorders in the presence of obesity and insulin resistance.
Control of hepatic gluconeogenesis
Prolonged fasting or starvation induces de novo glucose synthesis from non-carbohydrate precursors, termed hepatic gluconeogenesis. This process initiates from the conversion of pyruvate to oxaloacetate by pyruvate carboxylase (PC) in the mitochondria and eventually concludes in the conversion into glucose via several enzymatic processes in the cytosol.7, 8, 9 Among the substrates for gluconeogenesis are amino acids, which can be converted into either pyruvate or intermediates of the tricarboxylic acid cycle; lactate, which can be converted into pyruvate by lactate dehydrogenase; and glycerol (from increased lipolysis in the white adipocytes under fasting), which can be converted into dihydroxyacetone phosphate, a gluconeogenic intermediate (a two-step process that is catalyzed by glycerol kinase and glycerol 3-phosphate dehydrogenase). Key regulatory enzymes in that pathway, including glucose 6-phosphatase (G6Pase), fructose 1,6-bisphosphatase (Fbpase1), PC, and phosphoenolpyruvate carboxykinase (PEPCK), are activated under fasting conditions to enhance gluconeogenic flux in that setting.
Mitochondrial acetyl-CoA (derived from the increased fatty acid oxidation under fasting) functions as a key allosteric activator of PC, leading to the increased production of oxaloacetate for the gluconeogenesis. In addition, F26BP, which is a key allosteric regulator for glycolysis by activating PFK-1, was shown to inhibit gluconeogenesis via the allosteric inhibition of Fbpase1, which helps reciprocally control gluconeogenesis and glycolysis under different dietary statuses. Because Fbpase2 is activated but PFK-2 is inhibited under fasting, the lack of F26BP enables the activation of Fbpase1 and the increased production of fructose 6-phosphate in gluconeogenesis. The chronic activation of gluconeogenesis is ultimately achieved via transcriptional mechanisms. Major transcriptional factors that are shown to induce gluconeogenic genes include CREB, FoxO1, and several nuclear receptors (Figure 3).31
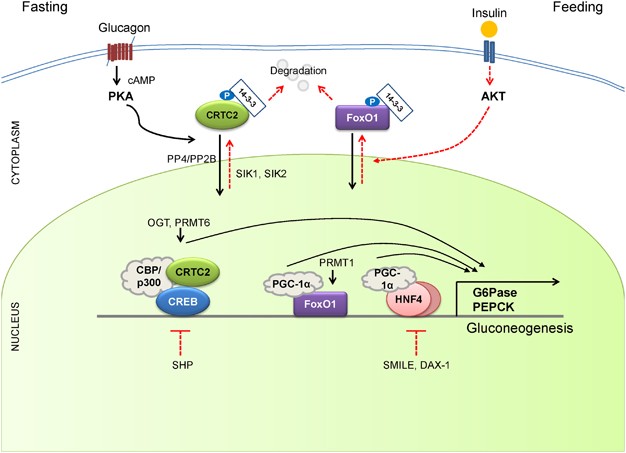
Regulation of hepatic gluconeogenesis. Under fasting conditions, hepatic gluconeogenesis is enhanced via a decreased concentration of insulin and an increased concentration of insulin counterregulatory hormones such as glucagon. CREB/CRTC2, FoxO1, and a family of nuclear receptors are critical in coordinating the fasting-mediated activation of gluconeogenesis in the liver. See the main text for more specific regulatory pathways. FoxO1, forkhead box O 1
Under fasting conditions, glucagon and epinephrine can increase the cAMP concentration in the liver via the activation of adenylate cyclase, leading to the activation of PKA and the subsequent induction of CREB via its serine 133 phosphorylation. The phosphorylation event is crucial in the recruitment of histone acetyltransferases (HAT) CBP/p300, leading to the histone H3 and H4 acetylation and the transcriptional activation of target genes.32, 33 CREB-dependent transcription is further enhanced by association with additional transcriptional coactivators CREB regulated transcription coactivators (CRTCs), which are a target for CBP/p300-mediated acetylation, which in turn promotes a tighter association of CREB, CBP/p300, and CRTC on the promoter.34, 35, 36 The role of CREB in the control of hepatic gluconeogenesis has been confirmed by in vivo studies by utilizing albumin promoter-driven ACREB (CREB inhibitor) transgenic mice and siRNA-mediated CREB knockdown mouse models.37, 38 In both mouse models, the inactivation of CREB reduced blood glucose levels and reduced the expression of gluconeogenic genes in mice, showing that CREB is a bona fide physiological transcriptional regulator of hepatic gluconeogenesis in vivo. In contrast, the role for CBP in gluconeogenesis is still controversial. Disruption of CREB-CBP interaction does not appear to affect glucose homeostasis because mice exhibiting a stable expression of mutant CBP that was unable to bind CREB showed normal glycemia.36 Furthermore, mutant mice producing CH1 null products (ΔCH1-a domain that is critical for insulin-mediated depression of CBP activity) displayed normal fasting gluconeogenesis.39 Thus, further studies are required to describe the potential role of HATs in the transcriptional control of CREB activity in this setting.
The CRTC family of transcriptional coactivators consists of CRTC1, CRTC2 and CRTC3, which were isolated by the expression library screening as activaters of CREB-dependent transcription.34 CRTC activity is regulated by cellular localization, and the AMPK family of serine/threonine kinases, such as AMPK, SIK1 or SIK2, was shown to be involved in the inhibitory phosphorylation of this factor (serine 171 for CRTC2).40 In addition, the phosphorylation status of CRTC is regulated by a pair of serine/threonine phosphatases (PP2B or PP4) in response to cAMP signaling or calcium concentration in the cell.41, 42 CRTC activity is also further enhanced by O-GlcNAcylation on serine 171 and arginine methylation by protein arginine methyltransferase (PRMT) 6.43, 44 Among the family members, CRTC2 is the prominent isoform in the liver. Recent studies have delineated the role of CRTC2 in the regulation of hepatic gluconeogenesis in vivo. Knockdown of CRTC2 in mice by RNAi reduced blood glucose levels and led to a concomitant repression of gluconeogenic gene expression.36 In addition, CRTC2 knockout mice displayed lower plasma glucose levels and improved glucose tolerance, indeed showing that CRTC2 is crucial in controlling hepatic glucose metabolism in vivo.45 A recent study indicated that CRTC2 could also coactivate other bZIP transcription factors that are implicated in the regulation of glucose homeostasis.46, 47 Further study is required to delineate the potential contributions from other bZIP factors in the control of hepatic gluconeogenesis by using tissue-specific knockout mouse models.
The forkhead box O (FoxOs) belongs to a class of forkhead families of transcription, which recognize the AT-rich insulin response element on the promoter.48, 49 Of the four major isoforms in mammals (FoxO1, FoxO3, FoxO4, and FoxO6), FoxO1 is the predominant isoform in the liver. The activity of this protein is also regulated by phosphorylation-dependent subcellular localization, and three major serine and threonine residues (threonine 24, serine 253 and serine 316 for murine FoxO1) are targeted by the insulin/Akt pathway. Following phosphorylation, FoxO1 moves to the cytosol via an association with 14-3-3, where it is degraded by the ubiquitin/proteasome-dependent pathway.50, 51, 52 In addition to phosphorylation, FoxO1 was shown to be regulated by the HAT-dependent acetylation of specific lysine residues (lysine 242, 245 and 262 for murine FoxO1), which also inhibit its transcriptional activity.53 In the liver, FoxO1 regulates hepatic gluconeogenesis via the transcriptional regulation of key genes in the pathway such as PEPCK and G6Pase and is considered a major regulatory point for the insulin-mediated repression of hepatic gluconeogenesis.54 Indeed, mice with liver-specific knockout of FoxO1 showed lower plasma glucose levels that those associated with reduced hepatic glucose output, thus underscoring the physiological significance of this factor in the control of glucose homeostasis in vivo.54, 55 As in the case for CREB, FoxO1 requires transcriptional coactivators for optimal transcriptional activity.
Peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α), a known coactivator for nuclear receptors, functions as a key transcriptional coactivator for FoxO1 in hepatic gluconeogenesis.56 PGC-1α was originally isolated in brown adipocytes and was shown to control adaptive thermogenesis in response to cold shock in that setting.57 In the liver, the expression of PGC-1α is upregulated under fasting conditions via a CRTC2-CREB-dependent mechanism and is critical in maintaining prolonged gluconeogenesis under starvation by enhancing the transcriptional activity of FoxO1 as a coactivator.38, 57, 58 Indeed, the depletion of hepatic PGC-1α in mice results in lower fasting glucose levels with a concomitant reduction in hepatic gluconeogenesis, thus showing the physiological relevance of this coactivator in the control of glucose homeostasis.59, 60 As is the case for CRTC2, FoxO1 activity is enhanced by arginine methylation by PRMT. In this case, PRMT1 promotes the asymmetric dimethylation of arginine 248 and 250 in FoxO1, which blocks the binding of Akt and the subsequent Akt-mediated phosphorylation of the adjacent serine residue (serine 253), thus enhancing the nuclear localization of FoxO1.61 Consequently, the chromatin occupancy of FoxO1 onto the gluconeogenic promoter and gluconeogenesis itself are increased due to the PRMT1-dependent arginine methylation.62 Acute knockdown of hepatic PRMT1 in mice reduces FoxO1-mediated glucose production, confirming that PRMT1 is crucial in modulating FoxO1 activity and subsequent gluconeogenesis in the physiological context.
Nuclear receptors belong to the superfamily of transcription factors that possess two Cys2-His2 type zinc finger motifs as a DNA binding domain as well as both ligand-independent and ligand-dependent transactivation domains.63 The latter activation domain also contains a ligand-binding domain. Nuclear receptors can be classified into one of three subgroups based on their dimer-forming potential. Homodimeric nuclear receptors are also called cytosolic receptors because they reside in the cytosol and associate with molecular chaperones such as heat-shock proteins. On binding to the ligand, they form homodimers and translocate to the nucleus to bind a specific response element termed the hormone response element to elicit the ligand-dependent transcriptional response. Most of the steroid hormone receptors, such as the glucocorticoid receptor (GR), estrogen receptor (ER), and progesterone receptor (PR), belong to this subfamily. By contrast, heterodimeric nuclear receptors reside in the nucleus and are bound to their cognate binding sites together with the universal binding partner retinoid X receptor (RXR). In the absence of the ligands, these factors repress the transcription of target genes in association with transcriptional corepressors such as histone deacetylase or nuclear receptor corepressor (NCoR)/silencing mediator of retinoid and thyroid hormone receptors (SMRT). Ligand binding initiates the conformational changes of these heterodimeric nuclear receptors, which promotes the dissociation of corepressors and the association of coactivators such as CBP/p300, p160 steroid receptor coactivator family, and PGC-1α.
Examples of this class of nuclear receptors include members of peroxisome proliferator-activated receptors, LXRs, vitamin D receptors and thyroid hormone receptors. The final subclasses of nuclear receptors are types that function as monomers. They usually lack specific endogenous ligands and are often called orphan nuclear receptors. Some of them also lack DNA binding domain and thus function as transcriptional repressors of various transcription factors, including members of nuclear receptors. They are called atypical orphan nuclear receptors. Among the homodimeric nuclear receptors, the role of GR has been linked to the control of hepatic gluconeogenesis. GR is activated by cortisol, which is released from the adrenal cortex in response to chronic stresses such as prolonged fasting.64, 65 GR was shown to directly bind to the cognate binding sites found in the promoters of gluconeogenic genes such as PEPCK and G6Pase and to enhance transcription of these genes under fasting conditions. The same response elements were also shown to be recognized and regulated by hepatocyte nuclear factor 4 (HNF4), a member of heterodimeric nuclear receptors, which suggests that these nuclear receptors could coordinately function to control hepatic gluconeogenesis in response to fasting.58
In accordance with this idea, the activity of these nuclear receptors can be effectively integrated by the function of transcriptional co-activator PGC-1α. Recently, estrogen-related receptor gamma (ERRγ), a member of monomeric nuclear receptors, was shown to be involved in the regulation of hepatic gluconeogenesis.66, 67 In the liver, ERRγ expression is increased under fasting or by insulin resistance in a CRTC2-CREB-dependent manner. This factor regulates hepatic gluconeogenesis by binding to unique response elements that are distinct from the known nuclear receptor-binding sites in the promoters of PEPCK and G6Pase. Inhibition of ERRγ activity by injecting either RNAi or the inverse agonist GSK5182 effectively reduced hyperglycemia in diabetic mice, suggesting that the control of this factor might potentially be beneficial in the treatment of patients with metabolic diseases. As is the case for other nuclear receptors that control hepatic gluconeogenesis, ERRγ activity is further enhanced by interaction with the transcriptional coactivator PGC-1α, showing that this coactivator functions as a master regulator for the hepatic glucose metabolism.
Three members of atypical orphan nuclear receptors, the small heterodimer partner (SHP, also known as NR0B2); the dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X (DAX-1, also known as NR0B1); and the SHP-interacting leucine zipper protein (SMILE) are implicated in the transcriptional repression of hepatic gluconeogenesis.68, 69, 70 SHP is ubiquitously expressed in mammalian tissues, with the highest expression occurring in the liver. Interestingly, metformin directly activates the transcription of SHP via an AMPK-mediated pathway. SHP directly inhibits cAMP-dependent transcription by binding to CREB, resulting in the reduced association of CREB with CRTC2.71, 72 The adenovirus-mediated overexpression of SHP could effectively reduce blood glucose levels in diabetic mice, thus showing the importance of this pathway in the control of hepatic glucose metabolism.
These results provide a dual mechanism for a metformin-AMPK dependent pathway to inhibit hepatic gluconeogenesis at the transcriptional level; an acute regulation of CRTC2 phosphorylation to inhibit the CRTC2-CREB-dependent transcriptional circuit; and a longer-term regulation of gluconeogenic transcription by enhanced SHP expression. Both DAX-1 and SMILE were shown to repress hepatic gluconeogenesis by inhibiting HNF4-dependent transcriptional events.73, 74 SIK1, a member of the AMPK-related kinases, was shown to enhance DAX-1 expression in the liver, whereas Akt was shown to activate the transcription of SMILE to target the HNF4 pathway under feeding conditions. Interestingly, SMILE was shown to directly replace PGC-1α from HNF4 and the gluconeogenic promoters, suggesting that this factor could potentially function as a major transcriptional repressor of hepatic gluconeogenesis in response to insulin signaling. Further study is necessary to fully understand the relative contribution of these nuclear receptors in the control of glucose homeostasis in both physiological conditions and pathological settings.
Concluding remarks
In this review, we attempted to describe the current understanding of the regulation of glucose metabolism in the mammalian liver. Under feeding conditions, glucose, a major hexose monomer of dietary carbohydrate, is taken up in the liver and oxidized via glycolysis. The excess glucose that is not utilized as an immediate fuel for energy is stored initially as glycogen and is later converted into triacylglycerols via lipogenesis. Glycogenesis is activated via the insulin-Akt-mediated inactivation of GSK-3, leading to the activation of glycogen synthase and the increased glycogen stores in the liver. Insulin is also critical in the activation of PP1, which functions to dephosphorylate and activate glycogen synthase. In addition, PP1 inhibits glycogenolysis via the dephosphorylation/inactivation of glycogen phosphorylase. Glycolysis is controlled by the regulation of three rate-limiting enzymes: GK, PFK-1 and L-PK. The activities of these enzymes are acutely regulated by allosteric regulators such as ATP, AMP, and F26BP but are also controlled at the transcription level. Two prominent transcription factors are SREBP-1c and ChREBP, which regulate not only the aforementioned glycolytic enzyme genes but also the genes encoding enzymes for fatty acid biosynthesis and triacylglycerol synthesis (collectively termed as lipogenesis).
The importance of these transcription factors in the control of glycolysis and fatty acid biosynthesis has been verified by knockout mouse studies, as described in the main text. The liver also has a critical role in controlling glucose homeostasis under fasting conditions. Initially, insulin counterregulatory hormones such as glucagon and epinephrine are critical in activating the PKA-driven kinase cascades that promote glycogen phosphorylase and glycogenolysis in the liver, thus enabling this tissue to provide enough fuel for peripheral tissues such as the brain, red blood cells and muscles. Subsequently, these hormones together with adrenal cortisol are crucial in initiating the transcriptional activation of gluconeogenesis such as PC, PEPCK and G6Pase. The major transcription factors involved in the pathway include CREB, FoxO1 and members of nuclear receptors, with aid from transcriptional coactivators such as CRTC, PGC-1α and PRMTs. These adaptive responses are critical for maintaining glucose homeostasis in times of starvation in mammals. Further study is necessary by using liver-specific knockout mice for each regulator of hepatic glucose metabolism to provide better insights into the intricate control mechanisms of glucose homeostasis in mammals.
References
- 1
Nordlie RC, Foster JD, Lange AJ . Regulation of glucose production by the liver. Annul Rev Nutr 1999; 19: 379–406.
- 2
Towle HC, Kaytor EN, Shih HM . Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu Rev Nutr 1997; 17: 405–433.
- 3
Roach PJ . Glycogen and its metabolism. Curr Mol Med 2002; 2: 101–120.
- 4
van de Werve G, Jeanrenaud B . Liver glycogen metabolism: an overview. Diabetes Metab Rev 1987; 3: 47–78.
- 5
Ros S, Garcia-Rocha M, Dominguez J, Ferrer JC, Guinovart JJ . Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. The J Biol Chem 2009; 284: 6370–6378.
- 6
Agius L . Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med 2015; 46: 34–45.
- 7
Pilkis SJ, Claus TH . Hepatic gluconeogenesis/glycolysis: regulation and structure/function relationships of substrate cycle enzymes. Annu Rev Nutr 1991; 11: 465–515.
- 8
Pilkis SJ, Claus TH, el-Maghrabi MR . The role of cyclic AMP in rapid and long-term regulation of gluconeogenesis and glycolysis. Adv Second Messenger Phosphoprotein Res 1988; 22: 175–191.
- 9
Pilkis SJ, el-Maghrabi MR, Claus TH . Hormonal regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Biochem 1988; 57: 755–783.
- 10
Brouwers MC, Jacobs C, Bast A, Stehouwer CD, Schaper NC . Modulation of glucokinase regulatory protein: a double-edged sword? Trends Mol Med 2015; 21: 583–594.
- 11
Dentin R, Girard J, Postic C . Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 2005; 87: 81–86.
- 12
Horton JD, Goldstein JL, Brown MS . SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109: 1125–1131.
- 13
Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem 1999; 274: 35832–35839.
- 14
Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab 2011; 13: 540–549.
- 15
Jeon TI, Osborne TF . SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 2012; 23: 65–72.
- 16
Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 2000; 14: 2819–2830.
- 17
Lu M, Shyy JY . Sterol regulatory element-binding protein 1 is negatively modulated by PKA phosphorylation. Am J Physiol Cell Physiol 2006; 290: C1477–C1486.
- 18
Bengoechea-Alonso MT, Ericsson J . A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem 2009; 284: 5885–5895.
- 19
Yoon YS, Seo WY, Lee MW, Kim ST, Koo SH . Salt-inducible kinase regulates hepatic lipogenesis by controlling SREBP-1c phosphorylation. J Biol Chem 2009; 284: 10446–10452.
- 20
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 2011; 13: 376–388.
- 21
Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci USA 2001; 98: 9116–9121.
- 22
Ma L, Tsatsos NG, Towle HC . Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J Biol Chem 2005; 280: 12019–12027.
- 23
Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V et al. The nuclear receptor LXR is a glucose sensor. Nature 2007; 445: 219–223.
- 24
Denechaud PD, Bossard P, Lobaccaro JM, Millatt L, Staels B, Girard J et al. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J Clin Invest 2008; 118: 956–964.
- 25
Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K . Mechanism for fatty acid "sparing" effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J Biol Chem 2002; 277: 3829–3835.
- 26
Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K . Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci USA 2001; 98: 13710–13715.
- 27
Tsatsos NG, Davies MN, O'Callaghan BL, Towle HC . Identification and function of phosphorylation in the glucose-regulated transcription factor ChREBP. Biochem J 2008; 411: 261–270.
- 28
Tsatsos NG, Towle HC . Glucose activation of ChREBP in hepatocytes occurs via a two-step mechanism. Biochem Biophys Res Commun 2006; 340: 449–456.
- 29
Iizuka K, Horikawa Y . ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr J 2008; 55: 617–624.
- 30
Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006; 55: 2159–2170.
- 31
Oh KJ, Han HS, Kim MJ, Koo SH . CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep 2013; 46: 567–574.
- 32
Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M et al. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature 1994; 370: 226–229.
- 33
Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH . Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993; 365: 855–859.
- 34
Altarejos JY, Montminy M . CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 2011; 12: 141–151.
- 35
Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB et al. TORCs: transducers of regulated CREB activity. Mol Cell 2003; 12 (2): 413–423.
- 36
Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005; 437: 1109–1111.
- 37
Erion DM, Ignatova ID, Yonemitsu S, Nagai Y, Chatterjee P, Weismann D et al. Prevention of hepatic steatosis and hepatic insulin resistance by knockdown of cAMP response element-binding protein. Cell Metab 2009; 10: 499–506.
- 38
Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001; 413: 179–183.
- 39
Bedford DC, Kasper LH, Wang R, Chang Y, Green DR, Brindle PK . Disrupting the CH1 domain structure in the acetyltransferases CBP and p300 results in lean mice with increased metabolic control. Cell Metab 2011; 14: 219–230.
- 40
Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 2004; 119: 61–74.
- 41
Wang Y, Li G, Goode J, Paz JC, Ouyang K, Screaton R et al. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 2012; 485: 128–132.
- 42
Yoon YS, Lee MW, Ryu D, Kim JH, Ma H, Seo WY et al. Suppressor of MEK null (SMEK)/protein phosphatase 4 catalytic subunit (PP4C) is a key regulator of hepatic gluconeogenesis. Proc Natl Acad Sci USA a 2010; 107: 17704–17709.
- 43
Dentin R, Hedrick S, Xie J, Yates J 3rd, Montminy M . Hepatic glucose sensing via the CREB coactivator CRTC2. Science 2008; 319: 1402–1405.
- 44
Han HS, Jung CY, Yoon YS, Choi S, Choi D, Kang G et al. Arginine methylation of CRTC2 is critical in the transcriptional control of hepatic glucose metabolism. Sci Signal 2014; 7: ra19.
- 45
Wang Y, Inoue H, Ravnskjaer K, Viste K, Miller N, Liu Y et al. Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci USA 2010; 107: 3087–3092.
- 46
Lee MW, Chanda D, Yang J, Oh H, Kim SS, Yoon YS et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab 2010; 11: 331–339.
- 47
Wang Y, Vera L, Fischer WH, Montminy M . The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 2009; 460: 534–537.
- 48
Accili D, Arden KC . FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004; 117: 421–426.
- 49
Barthel A, Schmoll D, Unterman TG . FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab 2005; 16: 183–189.
- 50
Biggs WH 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC . Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA 1999; 96: 7421–7426.
- 51
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96: 857–868.
- 52
Nakae J, Park BC, Accili D . Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem 1999; 274: 15982–15985.
- 53
Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci USA 2004; 101: 10042–10047.
- 54
Haeusler RA, Kaestner KH, Accili D . FoxOs function synergistically to promote glucose production. J Biol Chem 2010; 285: 35245–35248.
- 55
Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D . Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 2007; 6: 208–216.
- 56
Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003; 423: 550–555.
- 57
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM . A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998; 92: 829–839.
- 58
Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001; 413: 131–138.
- 59
Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R et al. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med 2004; 10: 530–534.
- 60
Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004; 119: 121–135.
- 61
Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell 2008; 32: 221–231.
- 62
Choi D, Oh KJ, Han HS, Yoon YS, Jung CY, Kim ST et al. Protein arginine methyltransferase 1 regulates hepatic glucose production in a FoxO1-dependent manner. Hepatology 2012; 56: 1546–1556.
- 63
Evans RM, Mangelsdorf DJ . Nuclear Receptors, RXR, and the Big Bang. Cell 2014; 157: 255–266.
- 64
van Schaftingen E, Gerin I . The glucose-6-phosphatase system. Biochem J 2002; 362: 513–532.
- 65
Zinker B, Mika A, Nguyen P, Wilcox D, Ohman L, von Geldern TW et al. Liver-selective glucocorticoid receptor antagonism decreases glucose production and increases glucose disposal, ameliorating insulin resistance. Metabolism 2007; 56: 380–387.
- 66
Kim DK, Gang GT, Ryu D, Koh M, Kim YN, Kim SS et al. Inverse agonist of nuclear receptor ERRgamma mediates antidiabetic effect through inhibition of hepatic gluconeogenesis. Diabetes 2013; 62: 3093–3102.
- 67
Kim DK, Ryu D, Koh M, Lee MW, Lim D, Kim MJ et al. Orphan nuclear receptor estrogen-related receptor gamma (ERRgamma) is key regulator of hepatic gluconeogenesis. J Biol Chem 2012; 287: 21628–21639.
- 68
Iyer AK, McCabe ER . Molecular mechanisms of DAX1 action. Mol Genet Metab 2004; 83: 60–73.
- 69
Seol W, Choi HS, Moore DD . An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science 1996; 272: 1336–1339.
- 70
Xie YB, Lee OH, Nedumaran B, Seong HA, Lee KM, Ha H et al. SMILE, a new orphan nuclear receptor SHP-interacting protein, regulates SHP-repressed estrogen receptor transactivation. Biochem J 2008; 416: 463–473.
- 71
Kim YD, Park KG, Lee YS, Park YY, Kim DK, Nedumaran B et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 2008; 57: 306–314.
- 72
Lee JM, Seo WY, Song KH, Chanda D, Kim YD, Kim DK et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J Biol Chem 2010; 285: 32182–32191.
- 73
Nedumaran B, Hong S, Xie YB, Kim YH, Seo WY, Lee MW et al. DAX-1 acts as a novel corepressor of orphan nuclear receptor HNF4alpha and negatively regulates gluconeogenic enzyme gene expression. J Biol Chem 2009; 284: 27511–27523.
- 74
Lee JM, Seo WY, Han HS, Oh KJ, Lee YS, Kim DK et al. Insulin-inducible SMILE inhibits hepatic gluconeogenesis. Diabetes 2016; 65: 62–73.
Acknowledgements
This work is supported by the National Research Foundation of Korea (grant nos.: NRF-2012M3A9B6055345, NRF-2015R1A5A1009024 and NRF- 2015R1A2A1A01006687), funded by the Ministry of Science, ICT & Future Planning, Republic of Korea, a grant of the Korean Health technology R&D Project (grant no: HI13C1886), Ministry of Health & Welfare, Republic of Korea and a grant from Korea University, Seoul, Republic of Korea.
Author information
Affiliations
- Division of Life Sciences, College of Life Sciences & Biotechnology, Korea University, Seoul, 136-713, Korea
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/