
16.2: Reactions of the Citric Acid Cycle
- Page ID
- 41618


Introduction
The acetyl-CoA formed by the pyruvate dehydrogenase complex (PDC) now enters a cyclic, non-linear pathway called the citric acid cycle, tricarboxylic acid (TCA) cycle, or the Krebs cycle after Hans Krebs who discovered it. The cycle is shown in Figure \(\PageIndex{1}\) in wedge/dash form with stereochemistry included to give a more exact representation.

Figure \(\PageIndex{1}\): Citric Acid Cycle
Why is this pathway a cycle and not a linear pathway as we have seen for glycolysis? A simple answer is that it evolved that way, but why would that be advantageous? It turns out that some of the key "intermediates" in the pathway are pulled away for the biosynthesis of other biomolecules. If the citric acid cycle was linear, and intermediates pulled off for other reactions, the linear pathway would taper off, which would not be optimal for a key energy production pathway. Of course, the removal of intermediates from a cyclic pathway would also slow it down but when this happens, enzymes outside of the cycle are used to produce key reaction intermediates of the cycle to it going. The replenishing reactions are called anaplerotic reactions.
Krebs, in his detailed analysis of the enzymes involved in "intermediary" metabolism, used radioisotope-labeled reactants to trace carbon atoms in respiring tissue. He found that when radiolabeled pyruvate and oxaloacetate were added to muscle tissue in vitro, radiolabeled citrate was formed.
Pyruvate + Oxaloacetate → citrate + CO2
This is correct but omits the initial conversion of pyruvate to acetyl-CoA. Hence the "end product" of the pathway (oxaloacetate) reforms the beginning reactant (citrate) so he surmised that the pathway was circular.
The next obvious question is why are eight reactions are required for the oxidation of just 2 Cs in pyruvate. Partly this is a matter of evolution again, as the early evolutionary pathway probably combined an oxidative (clockwise) set of reactions with a reductive (counterclockwise) set. Part of the chemistry in the cycle is devoted to producing either β-keto acids, which are easy to oxidatively decarboxylate, or converting α-ketoacid to molecules with better electron sinks β to the departing CO2. After the net 2 carbon atoms added to the cycle are released as 2 CO2s, the rest of the reactions are used to regenerate oxaloacetate, allowing the cycle to continue.
Of course, the ultimate goal of an energy-extractive oxidative pathway is not just to form CO2 but to form ATP or its equivalent (i.e. GTP). Notice that 3 NAD+s are used and converted to 3 NADH. In addition, a new, more potent oxidizing agent, FAD, is used and it is converted to FADH2. NAD+ and FAD are replenished by reoxidation of NADH and FADH2 (reduced forms) back to NAD+ and FAD, through mitochondrial electron transport (oxidation) reactions, in which electrons are passed to stronger and stronger oxidizing agents, the last being O2. In this thermodynamically favored process, lots of ATPs are made. We will explore those reactions in the next section.
We will go through each of the steps in the citric acid cycle separately and show how the pathway is regulated (section 16.3). Why such detail? There are only 8 steps. It seems that we should be able to carefully examine each given that the citric acid cycle is a hub that along with glycolysis controls metabolic flow through many interconnected metabolic pathways.
At the same time, we can't explore each reaction in every pathway described in this text in great detail, otherwise, this book would become more of an encyclopedia. In this chapter and beyond, we will focus on mechanistic details only of enzymes that catalyze different types of reactions than those in glycolysis or the citric acid cycle, and those with interesting cofactors and mechanisms.
Other issues add complexity for learners. The PDC and citric acid cycle reaction occur in the mitochondrial matrix. Cytoplasmic pyruvate and NAD+ must be transported into the matrix from the cytoplasm. In addition, some of the enzymes in the citric acid cycle have both cytoplasmic and mitochondrial variants. Some of these homologous pairs are differentiated by their use of NAD+ or NADP+ as an oxidizing agent. The ones in the cytoplasm are not part of the cycle. You would expect these enzyme pairs to have similar tertiary structures and active site chemistry. Prokaryotic forms of these enzymes are similar structurally to their eukaryotic forms so the interactive molecular models shown below will show enzymes from a variety of organisms. Finally, there are many variants, shunts, and bypasses of the citric acid pathway in different organisms. We will explore this topic in section 16.4
We will try to pair reaction mechanism diagrams that show the flow of electrons in bond-making and breaking with interactive molecular models of the active site. You should rotate the models to align and identify key amino acids and ligands (substrate, substrate analogs, inhibitors, activators) shown in the static 2D mechanism diagrams. Since the active sites are often conserved across prokaryotic and eukaryotic versions, the choice of PDB structures used depends on which best illustrates a conceptual point.
There are many ways to write abbreviated chemical equations showing NAD+/NADH and FAD/FAD2 and hydrogen ions in metabolic pathway diagrams. To make sense of them, consider a simplified mechanism for the oxidation of ethanol by alcohol dehydrogenase, as shown in Figure \(\PageIndex{2}\). Note that there are 2 Hs on the oxidized substrate (ethanol) that are involved. One is a hydride and the other is a proton.

Figure \(\PageIndex{2}\): Oxidation of ethanol using NAD+
Here is a list of different and seemingly contradictory ways to write a chemical equation to show changes in NAD+/NADH and H ions:
- NAD+ + :H- → NADH. This chemical equation is charge balanced and shows just the changes to the NAD+/NADH pair, but it doesn't show the proton (H+) lost from the substrate.
- NAD+ + 2e- + H+ → NADH. This is the same as equation (1) but with the hydride separated into an electron pair and a proton.
- NAD+ + H+ → NADH. This is balanced for Hs but not + charge as it doesn't explicitly show the electron pair from the hydride added to NAD+.
- NAD+ → NADH + H+. This is balanced for charge but not for Hs. The extra H+ is the proton from the oxidized substrate.
We will use example 4 above throughout this book. That equation is most useful when trying to account for the change in the number of protons in the individual reactions and entire pathways. We will also write simplified chemical equations involving FAD (in which 2H from the substrate are added) as FAD → FADH2.
1. Citrate Synthase (CS)
Oxaloacetate + Acetyl-CoASH + H2O → Citrate + CoASH ΔGo = -7.5 kcal/mol (-31 kJ/mol)
Acetyl CoA is a thioester. Hence it is "high energy" compared to its hydrolysis products. (Remember, there is no such thing as a "high energy" bond.) The free energy released in its hydrolysis is used to drive the reaction forward. This is important otherwise citrate would not be formed readily. This reaction feeds the end product of glycolysis into the citric acid cycle. It is summarized in Figure \(\PageIndex{3}\).

Figure \(\PageIndex{3}\): Summary reaction - Citrate Synthase
The enzyme exists in two major conformations, an open and closed form. When the open form, which has a binding site for oxaloacetate, binds the substrate, a shift to the closed conformation forms on the binding site of acetyl-CoA. These changes sequester the bound substrates and exclude water and prevent spurious hydrolysis of acetyl-CoA. The binding occurs sequentially so the kinetics follow a sequential ordered mechanism.
Figure \(\PageIndex{4}\) left shows an animated gif that shows the conformational changes between the citrate-bound version (open, green) and the citrate and CoASH-bound form (blue). The image below right shows a smoother transition between the open and closed form without bound ligands (1cts, 2cts)
 |
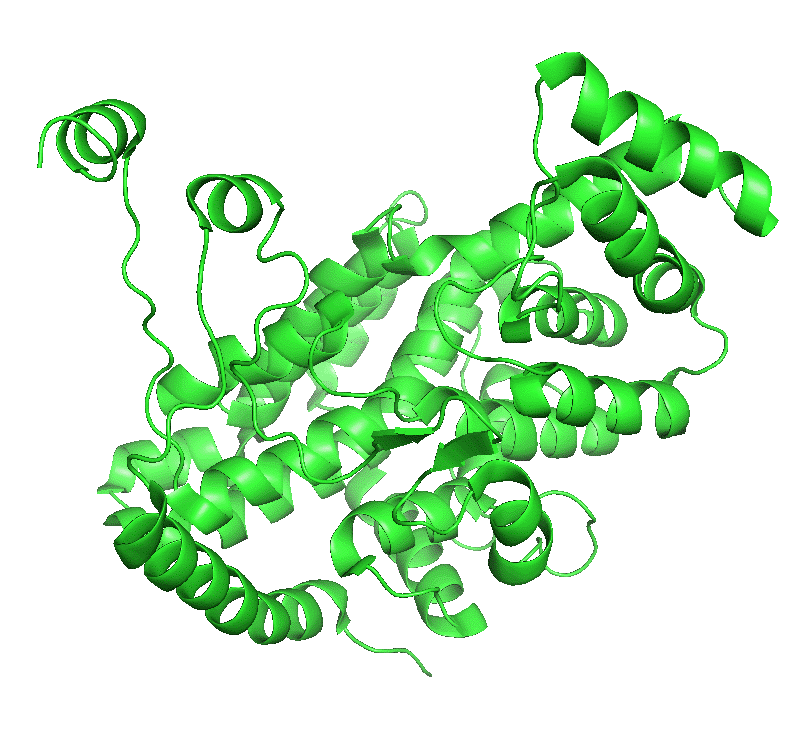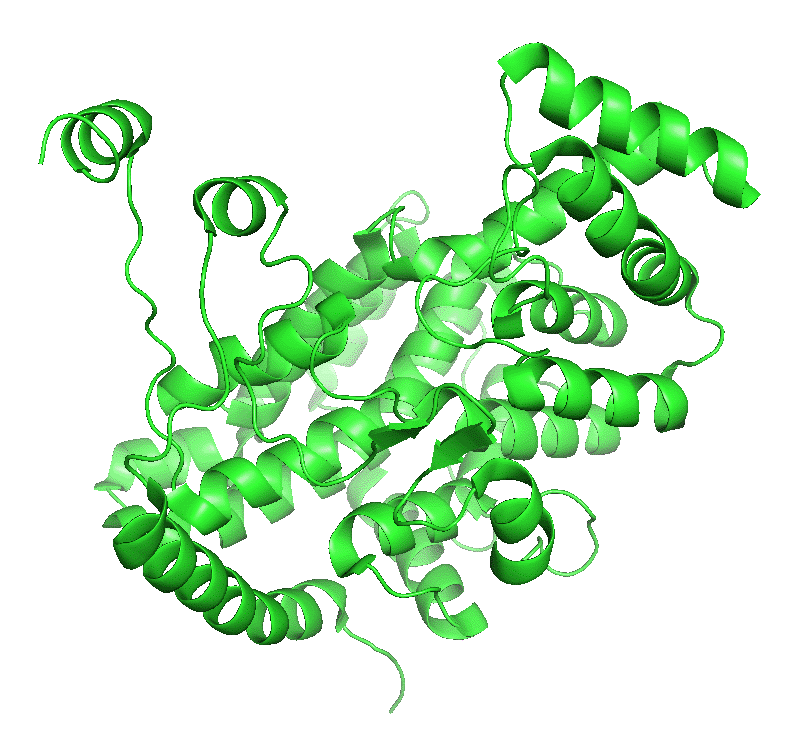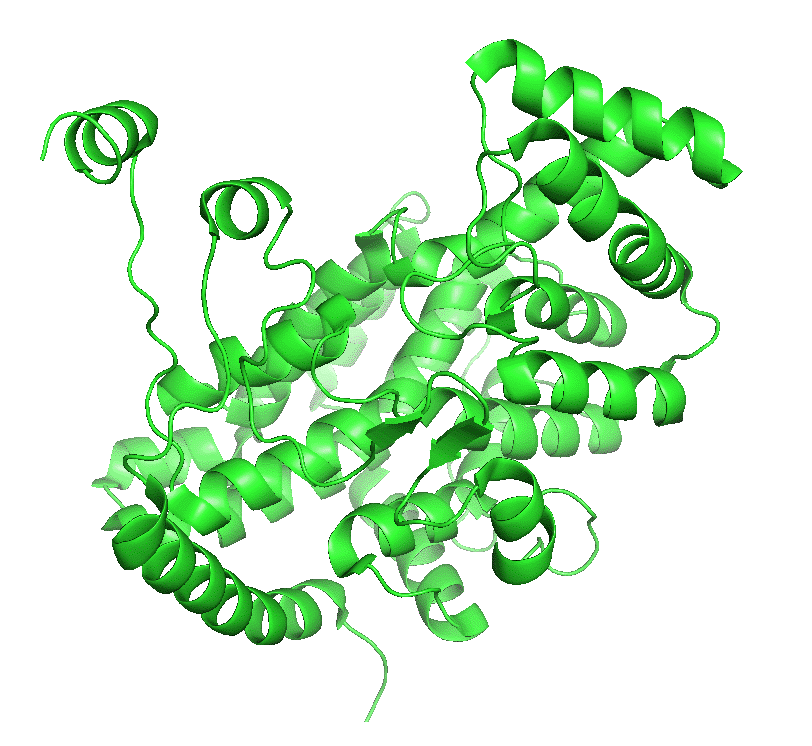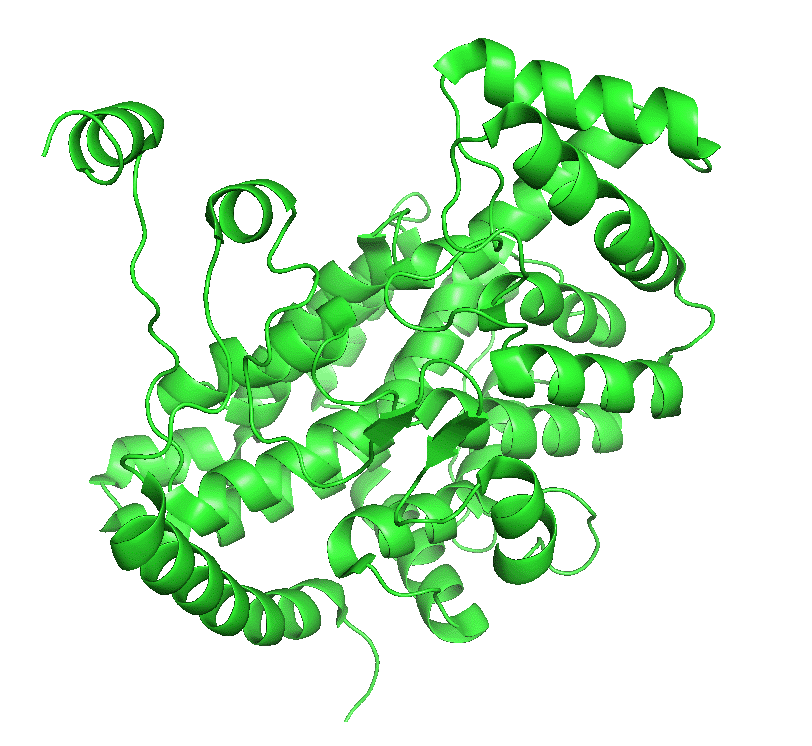 |
Figure \(\PageIndex{4}\): Conformational changes in citrate synthase on binding substrate
 For a more details view of the enzyme view the Regulation of Citrate Synthase.
For a more details view of the enzyme view the Regulation of Citrate Synthase.
The mechanism below is from https://chem.libretexts.org/Bookshel...trate_Synthase, with Contributors and Attributions from:
- Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
- Prof. Steven Farmer (Sonoma State University)
- Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)
In this reaction, a C-C bond must form between the substrates. One way to do that is to make a nucleophilic carbanion ion from the alpha carbon of acetyl CoA. Remember, this is not a decarboxylation reaction so we don't have to worry about an electron "sink" on the beta carbon. Forming the carbanion would be possible since the negative charge on the carbon can be withdrawn to the carbonyl oxygen to form an enolate. The enolate becomes even more stable if the negative oxygen is protonated. So this is reaction is an aldol condensation, the addition of an enolate to an aldehyde or ketone. The carboxylate group of aspartic acid 375 on citrate synthase removes the acidic alpha proton on acetyl CoA, while histidine 274 donates a proton to form the neutral enol, a much more stable molecule than the enolate anion. His 274 continues to stabilize the enol during the reaction. Bound oxaloacetate is stabilized in part by Arg 329. In the next part of the mechanism, a second histidine (320) protonates the carbonyl oxygen of oxaloacetate, activating the carbonyl carbon for nucleophilic attack by the enol in the next step to form (S)-citryl CoA. The hydrolysis of the CoASH occurs when His 320 deprotonates a water molecule, facilitating nucleophile attack on the carbonyl carbon bonded to -SCoA, forming citrate.
A plausible mechanism is shown in Figure \(\PageIndex{5}\).

Figure \(\PageIndex{5}\): Citrate synthase mechanism
Figure \(\PageIndex{6}\) shows an interactive iCn3D model of the pig citrate synthase bound to CoASH and citrate (2CTS)
.png?revision=1&size=bestfit&width=488&height=371)
 Figure \(\PageIndex{6}\): pig citrate synthase bound to CoASH and citrate (2CTS). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...WXqcEsu336tcL7
Figure \(\PageIndex{6}\): pig citrate synthase bound to CoASH and citrate (2CTS). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...WXqcEsu336tcL7
The enzyme is a dimer with monomers shown in different colors. Citrate and CoASH are shown in sticks and labeled. The active site residues are shown as sticks and labeled in each subunit.
2. Aconitase
Citrate ↔ Isocitrate ΔGo = +2 (rx 2a), -0.5 (rx 2b) kcal/mol; net ΔGo = + 1.5 kcal/mol (+6.3 kJ/mol)
Thinking like a chess player, who must anticipate future moves, the chemical rationale for this reaction is to move an OH to a beta position, which in a subsequent reaction is converted to a beta C=O, so it can act as an electron sink to facilitate decarboxylation in a following reaction! The reaction is readily reversible (note the low ΔGo value) since the reactant and product are simple isomers of each other. Figure \(\PageIndex{7}\) shows the summary reaction.

Figure \(\PageIndex{7}\): Summary reaction of aconitase
The enzyme has an inorganic Fe4S4 cluster. Each Fe in the cluster coordinates to 4 S2- in a cubane structure, but when either citrate or isocitrate is bound, one of the Fe ions interacts with both the Os of a substrate carboxylate shown. The other two carboxylates of isocitrate are stabilized through ion-ion interactions by Arg 446 and 663.
Figure \(\PageIndex{8}\): below shows a plausible partial mechanism. An active site deprotonated serine abstract a proton at the S carbon. This is followed by the formation of the C-C double bond and a release of the resulting cis-aconitate from one bond to the FeS cluster.

Figure \(\PageIndex{8}\): Mechanism of aconitase
Figure \(\PageIndex{9}\) shows an interactive iCn3D model of the bovine S642A aconitase with bound citrate (1C97).
.png?revision=1&size=bestfit&width=527&height=348)
Figure \(\PageIndex{9}\): Bovine S642A aconitase with bound citrate (1C97). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...iJJRGaFoCDnhf7
This cis-aconitate intermediate in the interconversion of citrate and isocitrate must do an 1800 flip around the C=C double bond. This is followed by rehydration to form the other isomer. The deprotonated His 101 abstracts a hydrogen from a water-bound to the FeS cluster, with the hydroxide acting as a nucleophile, which along with the redonation of a hydrogen ion on the protonated Ser 642 the alpha-carbon completes the rehydration step in the formation of the other isomer.
Exercise \(\PageIndex{1}\)
Why was the S642A mutant used to produce the structure shown in the above iCn3D model?
- Answer
-
It allows the binding of substrate/product, in this case, isocitrate, to an inactive enzyme as the active site serine was mutated to a non-nucleophilic alanine of similar size. Hence no bond-making/breaking occurs in the complex.
References:
3. Isocitrate Dehydrogenase (IDH)
Isocitrate + NAD+ → α-ketoglutarate + NADH + H+ ΔGo = -2.0 kcal/mol (-8.4 kJ/mol)
The chemical rationale should be clear. In this step, through an oxidative decarboxylation, the CO2 is removed as NADH is produced. The NADH will be reoxidized back to NAD+ in the electron transport chain, leading to ATP production (see next section). The reaction is summarized in Figure \(\PageIndex{10}\).

Figure \(\PageIndex{10}\): Summary reaction: isocitrate dehydrogenase
Exercise \(\PageIndex{1}\)
Why must the oxidation reaction precede the decarboxylation reaction?.
- Answer
-
First, a beta-ketoacid intermediate must form, which allows easy decarboxylation of the intermediate as the beta carbonyl provides an electron "sink" to facilitate the decarboxylation.
There are two forms of this enzyme (IDH), a cytoplasmic (NADP+) form and a mitochondrial (NAD+ ) form. The cytoplasmic forms from various organisms are homodimers and have a common mechanism of catalysis. In contrast yeast mitochondrial IDH has two subunits, IDH1 (regulatory, binds the allosteric activator citrate and AMP) and IDH2 (catalytic, binds isocitrate and NAD+). Mammalian IDHs are tetramers heterodimers (αβ + αγ), which can also form a heterooctamer (αβ + αγ)2. The alpha chain is the catalytic subunit.
Mammalian NAD-IDHs are even more complex than yeast NAD-IDH. These enzymes are composed of three types of subunits, α, β, and γ, which share about 40–52% sequence identity. The α and β form an αβ dimer, while α and γ subunits form αγ. These then interact to form the α2βγ heterotetramer, which effectively forms the holoenzyme. It can also form an active heterooctamer.e. The αγ heterodimer is regulated by citrate and ADP. On citrate binding to the allosteric site, a conformation change occurs to enhance isocitrate binding. ADP enhances the binding of the allosteric regulator citrate.
Figure \(\PageIndex{11}\) shows an interactive iCn3D model of that shows the superposition of the α chains of cytoplasmic IDH (NADP, sky blue, 4L03) and mitochondrial IDH (NAD) (6KDY, salmon).
_and_mitochondrial_IDH_(NAD)_(6KDY).png?revision=1&size=bestfit&width=345&height=396)
Figure \(\PageIndex{11}\): Superposition of the A chains of cytoplasmic IDH (4L03) and mitochondrial IDH (NAD) (6KDY) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...zy9yn4NaF9g1c9
Cytoplasmic IDH is sky blue and mitochondrial IDH is salmon. Click the 3 bar menu icon (top left) in the model in the window, scroll down to Alternate, and toggle back and forth between the two forms. The structures of the alpha chains, although not identical, align well.
Figure \(\PageIndex{12}\) shows a probable mechanism for the reaction based on the conserved catalytic site shown in the model above.

Figure \(\PageIndex{12}\): Mechanism of isocitrate dehydrogenase (after https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3706558/)
Figure \(\PageIndex{13}\) shows an interactive iCn3D model of active site of the cytoplasmic human IDH1 in complex with NADP+ and Ca2+/α-ketoglutarate (4L03).
.png?revision=1&size=bestfit&width=396&height=367)
Figure \(\PageIndex{13}\): Active site of the cytoplasmic human IDH1 in complex with NADP+ (NAP1) and Ca2+/ alpha-ketoglutarate (4L03) (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...P9DqLoPowEV2d9
Figure \(\PageIndex{14}\) shows an interactive iCn3D model of the active site of the αβ heterodimer of human IDH3 (6kdy) in complex with NAD+.
_in_complex_with_NAD.png?revision=1&size=bestfit&width=460&height=368)
Figure \(\PageIndex{14}\): αβ heterodimer of human IDH3 (6kdy) in complex with NAD+(6kdy) . (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...hyyJiMFEqwktB7
The α subunit is shown in gray and the β in cyan.
4. α-ketoglutarate dehydrogenase
α-ketoglutarate + NAD+ + CoASH → succinyl CoA + CO2 + NADH + H+ ΔGo = -7.2 kcal/mol (-30 kJ/mol)
In the last reaction, α-ketoglutarate was formed. Oh no, you might say! It would have been nice to form a β-ketoacid, which could easily decarboxylation. No worries though. We spent all of section 16.1 explaining the biochemistry used to decarboxylate another α-ketoacid, pyruvate. The same chemistry is used to accomplish the oxidative decarboxlation of α-ketoglutarate. Hence we won't expand on the mechanism here. The reaction is shown in Figure \(\PageIndex{15}\).

Figure \(\PageIndex{15}\): Summary reaction for α-ketoglutarate dehydrogenase
5. Succinyl-CoA synthetase (SCS)
succinyl CoA + GDP + Pi → succinate + CoASH + GTP ΔGo = -0.8 kcal/mol (-3.3 kJ/mol)
This is the first step in which the energy change in the cycle is captured specifically in the form of a high energy (with respect to its hydrolysis product) phosphoanhydride bond in the form of GTP (and ATP in some organisms). From a chemical step, the cleavage of the thermodynamically unstable thioester is coupled to the endergonic synthesis of GTP. This can transfer its terminal phosphate to ADP to make ATP in reaction that has a ΔGo of about 0 kcal/mol. The reaction is shown in Figure \(\PageIndex{16}\).

Figure \(\PageIndex{16}\): Summary reaction for succinyl-CoA synthetase
Succinyl-CoA synthases have two subunits, α and β. The enzyme in E. coli is a tetramer (α2β2) with the catalysis occurring at the αβ interface. The alpha-subunits interact only with the beta-subunits, whereas the beta-subunits interact to form the dimer of alpha beta-dimers with CoA bound in each α subunit to a nucleotide-binding loop.
Two histidines, His 246 and His 142 are involved in the reaction, with His 246 becoming phosphorylated to form an intermediate in the reaction. A mutation of His 142 to an asparagine (H142N) essentially abolishes enzyme activity. Different SCSs have different specificities for purine nucleoside triphosphates. Organisms, including mammals, may have two different isoforms, one that binds ADP and one that uses GDP (as shown in most diagrams of the citric acid cycle). In E. Coli, the α subunit binds CoASH and contains His 246, which gets phosphorylated. The β subunit determines the specificity for either GTP or ATP. In E.Coli the ATP binding site (Site II "in the ATP-grasp fold") is quite distant from the CoASH site (Site II) so phospho-His 246 must move between the sites in the dimer interface.
The three steps in the reaction are shown below, where E is the free enzyme, a . indicates a noncovalent complex, and a - represents a covalent bond (after Biochemistry 2002, 41, 537-546)
- E + succinyl-CoA + Pi ↔ E . succinyl-PO3 + CoASH
- E . succinyl-PO3 ↔ E-PO3 + succinate
- E-PO3 + NDP ↔ E + NTP
Figure \(\PageIndex{17}\) shows an abbreviated mechanism that shows only the involvement of His 246.

Figure \(\PageIndex{17}\): Abbreviated mechanism for succinyl-CoA synthase
Kinetic analysis suggests that the three substrates bind in a specific order, catalysis occurs, and then the three products leave. This type of reaction is called an ordered ter ter reaction, and is shown in Figure \(\PageIndex{18}\).

Figure \(\PageIndex{18}\): Ordered ter ter reaction for succinyl-CoA synthase
The iCn3D model below shows key alpha-chain residues in the active site, including the phosphorylated His 246 and bound CoASH. (use iCn3D to visualized 1CQJ, the nonphosphorylated form) I)
Figure \(\PageIndex{19}\) shows an interactive iCn3D model of the complex of ADP and Mg2+ with Dephosphorylated E. Coli Succinyl-CoA Synthetase (1CQI)
.png?revision=1&size=bestfit&width=443&height=456)
Figure \(\PageIndex{19}\): Complex of ADP and Mg2+ with Dephosphorylated E. Coli Succinyl-CoA Synthetase (1CQI). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...BssxsabV1S9cz6
Just one αβ dimer is shown. The α subunit is shown in gray and the β in cyan. Bound Pi and CoASH are labeled. The active site His246 in the α chain is shown in stick and labeled.
Figure \(\PageIndex{20}\) shows an interactive iCn3D model of the active site of pig GTP-specific succinyl-CoA synthetase in complex with succinate and CoASH (5CAE).
.png?revision=1&size=bestfit&width=519&height=337)
Figure \(\PageIndex{20}\): Active site of pig GTP-specific succinyl-CoA synthetase in complex with succinate and CoASH (5CAE). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...HjH9ER5wLqb1T7
6. Succinate Dehydrogenase
succinate + FAD ↔ fumarate + FADH2 ΔGo = 0 kcal/mol
The enzyme is yet another step in closing the cycle to reform oxaloacetate. It is also our first encounter with FAD as an oxidizing agent. Its reduction product, FADH2, will be reoxidized in the electron transport chain (mitochondrial inner membrane for eukaryotes), producing energy for ATP. Hence it can be considered a proxy ATP-generating reaction. The reaction is shown in Figure \(\PageIndex{21}\).

Figure \(\PageIndex{21}\): Summary reactoin for succinate dehydrogenase
The succinate dehydrogenase enzyme is part of the larger Complex II of the electron transport chain. Complex II has many cofactors involved in its overall activity. It also uses an iron/sulfur cluster cofactor, similar to aconitase, which also produces a C=C doubled bonded intermediate. We will discuss it in greater detail in the chapter on electron transport. For now, let's concentrate on this new cofactor and oxidizing agent, FAD. Many enzymes use FAD/FADH2 in redox chemistry.
In contrast to NAD+/NADH, the FAD/FADH2 pair stays tightly bound to the enzyme and doesn't readily dissociate. This means that after one cycle of the enzyme (after FAD is converted to FADH2), the enzyme is functionally "dead". Another oxidizing agent must bind to the enzyme and reoxidize FADH2 back to FAD. The dissociation constants for FAD/FADH2 and its protein binder in a flavoprotein are often in the nanomolar range. In around 10% of flavoproteins, FAD/FADH2 are usally covalently bonded to the enzyme.
Figure \(\PageIndex{22}\) shows an interactive iCn3D model of the Avian respiratory complex II FAD binding subunit with FAD and a malate-like intermediate (1YQ3).
.png?revision=1&size=bestfit&width=503&height=396)
Figure \(\PageIndex{22}\): Avian respiratory complex II FAD binding subunit with FAD and a malate-like intermediate (1YQ3). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...SGac3NuN4XBWk7
Note how buried the FAD is in the middle of the dehydrogenase subunit. The malate-like intermediate (TEO) is shown next to the FAD in yellow spacefill.
Exercise \(\PageIndex{1}\)
Succinate dehydrogenase is irreversibly inhibited by the toxin 3-nitroproprionic acid (3np) made by some plants and fungi. Eating moldy sugar cane has led to reported deaths.
1. Draw the Lewis structures of succinate and 3-nitroproprionic acid. Compare them and the total number of valence electrons in each.
2. Here is a link to an iCn3D model showing the interaction of 3np with the enzyme. Explain the mode of action of the toxin.
https://structure.ncbi.nlm.nih.gov/i...cja9VKU2ZMCac9
- Answer
-
These molecules are structurally similar and isoelectronic (the same number of electrons in their Lewis structures.
The inhibitor 3np form a covalent adduct through the guanidino group of Arg 297. This is a key catalytic residue that act as a general base that accepts a proton from succinate in the reaction.
Figure \(\PageIndex{23}\) (top) shows a very general and abbreviated mechanism for the enzyme for the immediate reaction of succinate with FAD. The bottom part of the image shows the amino acids surrounding succinate in the avian (bird) version of the enzyme (pdb 1yq4)

Figure \(\PageIndex{23}\): Top - a very general and abbreviated mechanism for the enzyme for the immediate reaction of succinate with FAD. Bottom - amino acids surrounding succinate in the avian (bird) version of the enzyme (1yq4)
Exercise \(\PageIndex{1}\)
One of the amino acids surrounding succinate in the figure above acts as a general base and abstracts a protein from succinate as a hydride is transferred (from a plane above) to FAD. Go to this iCn3D of the active site bound to FAD. Which amino acid is the likely general base? (Note: the figure below shows general protonated states of side chains and not necessarily those involved in the proton abstraction. Go to Analysis, Distance and Distance between 2 atoms to find the likely general base.
- Answer
-
Arg 297
7. fumarase
fumarate + H2O ↔ L-malate ΔGo = -0.9 kcal/mol (-3.8 kJ/mol)
The chemical rationale for this reaction is clear - it is the penultimate step in the resynthesis of oxaloacetate, one of the reactants that starts the cycle, allowing the cycle to continue. This reaction introduces an O by hydration which can be oxidized in next step to produce NADH for e- transport/ATP production. The reaction is shown in Figure \(\PageIndex{24}\).

Figure \(\PageIndex{24}\): Summary reaction for fumarase
There are Class I (dimers containing an unstable FeS cluster, examples A and B) and Class II (tetramer, no bound iron, oxygen stable, example C) fumarases. Humans have both cytoplasmic and mitochondrial type II fumarases, resulting from alternative transcription of the fumarase genes. We will consider the type II, fumarase C from E. Coli in the following discussion.
The tetramer contains just alpha helices and random coils and has two distinct binding sites. Site A appears to be the active site and contains a buried water molecule. Site A, formed from three of the subunits, binds competitive inhibitors such as citrate and β-(trimethylsilyl)maleate, a cis substrate for fumarase, and is buried. 12 Angstroms away is site B, which is found in only one of the subunits near a pi-helix (H129 through N135) and is more surface-exposed. Each site has a histidine, but mutation of only one H188N in the A site disrupts enzyme activity. Both sites bind multi-carboxylates. The role of site B is a bit unclear, but it is most likely an allosteric site involved in the transfer of product (malate) from the buried site to the surface for ultimate dissociation.
Figure \(\PageIndex{25}\) shows an interactive iCn3D model of the fumarase with beta-(trimethylsilyl)maleate and citrate (1fuq).
maleate_and_citrate_(1fuq).png?revision=1&size=bestfit&width=352&height=387)
Figure \(\PageIndex{25}\): Fumarase with beta-(trimethylsilyl)maleate and citrate (1fuq). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...MewEQFebi4VL48
The monomers in the tetramer are shown in different colors. Citrate (Cit) and beta-(trimethylsilyl)maleate (SIF) are shown in spacefill.
Figure \(\PageIndex{26}\) shows an interactive iCn3D model of the binding site of citrate, a competitive inhibitor, of fumarase (1FUQ).
.png?revision=1&size=bestfit&width=296&height=271)
Figure \(\PageIndex{26}\): Binding site of the competitive inhibitor citrate in fumarase (1FUQ). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...yfPc5iPWbExr86
Figure \(\PageIndex{27}\) shows a plausible mechanism for the trans addition of water to fumarate.

Figure \(\PageIndex{27}\): Mechanism for fumarase
8. Malate Dehydrogenase (MDH)
L-malate+ NAD+ ↔ oxaloacetate + NADH + H+ ΔGo = +7.1 kcal/mol (30 kJ/mol)
We are finally there! This last reaction of the citric acid cycle produces oxaloacetate, the starting reactant, so the cycle can continue. It also produces NADH for mitochondrial e- transport/ATP production. Notice that is thermodynamically unfavorable (in the standard state) but the reaction is pulled to citrate formation by the first and next step of the cycle, citrate synthase. The reaction is shown in Figure \(\PageIndex{28}\).

Figure \(\PageIndex{28}\): Summary reaction for malate dehydrogenase
Malate dehydrogenases are found in the cytoplasm, where it is part of the aspartate-malate shuttle that moves cytoplasmic malate (and through MDH indirectly NADH) into the mitochondria. It is also found in the mitochondria, where it is part of the citric acid cycle. There are also NAD+ and NADP+-dependent forms. Malate can undergo two different types of oxidation reactions, one producing oxaloacetate and using NAD+, and one, an oxidative decarboxylation producing pyruvate and CO2, using NADP+. The latter is sometimes called malic enzyme.
Humans have two forms (MDH 1 and MDH 2) that use NAD+. The enzyme is a homodimer in humans with binding sites on both. It activity is allosterically
regulated by citrate, and it is inhibited by many things, including ATP, ADP, AMP, fumarate, citrate, aspartate, and high concentrations of oxaloacetate.
The enzyme is similar to lactate dehydrogenase, which we encountered in the chapter of glycolysis. Kinetic analyses show that NAD+ binds first followed by malate.
Figure \(\PageIndex{29}\) shows an interactive iCn3D model of NAD+ and malate bound to human malate dehydrogenase 2 (4wlu).
.png?revision=1&size=bestfit&width=408&height=296)
Figure \(\PageIndex{29}\): NAD+ and malate bound to human malate dehydrogenase 2 (4wlu). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...DrAXBdu64GPzx8
Just one monomer is shown. NAD+ and malate (LMR) are shown in sticks and labeled.
Figure \(\PageIndex{30}\) shows an abbreviated mechanism for malate dehydrogenase. The numbers refers to the E. Coli enzyme.

Figure \(\PageIndex{30}\): Abbreviated mechanism for malate dehydrogenase
Note that the hydride transferred from the malate is shown in red as a deuterium (D). It is transferred to the re face of NAD+ to form NADH. The carbon with the transferred deuterium in NADH is prochiral. Think of that as that carbon being chiral if one of the 2 Hs could be arbitrarily assigned a higher priority in assigning R/S isomers. D has a higher priority than H in the Cahn/Ingold designation system. In the reverse reaction, the D atom, which is above the plane of the ring, occupies the proR position. The proR deuterium is transferred back in this reversible reaction. A D was used simply to indicate the stereochemistry and to assign it in NADH to the proR position.
Figure \(\PageIndex{31}\) shows an interactive iCn3D model of the active site of the E. Coli malate dehydrogenase with bound citrate and NAD+ (1EMD).
.png?revision=1&size=bestfit&width=504&height=366)
Figure \(\PageIndex{31}\): Active site of the E. Coli malate dehydrogenase with bound citrate and NAD+ (1EMD). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...mHhmeFQyPpZj18
We have shown many renderings of the enzymes involved in the cycle. Yet another one is shown below. Figure \(\PageIndex{32}\) shows an interactive iCn3D model of the electrostatic surface potential of malate dehydrogenase (4WLU).
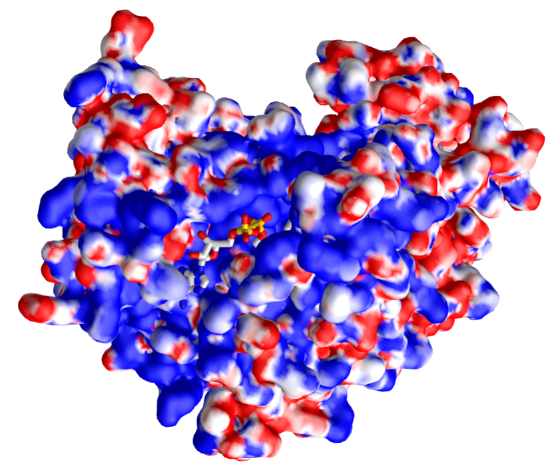
Figure \(\PageIndex{32}\): Electrostatic surface potential of malate dehydrogenase (4WLU)with bound NAD+ (4WLU). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/icn3d/share.html?izf2dsWXTw2bE8YD7
The display surface is the electrostatic surface potential map of the enzyme. Red shows the surfaces that are more anionic and with a negative electrostatic potential to which cationic molecules would be attracted, while blue represents more cationic surfaces to which the anion would be attracted. Note that the bound NAD+, which has many oxygens which are slightly or fully negative, is bound in a blue, positive electrostatic potential region.
Summary
Let's do some stoichiometry for the full cycle. Here is the net reaction (assuming that the GTP produced by succinyl-CoA synthetase is equivalent to 1 ATP).
Acetyl-CoA + 3NAD+ + FAD + ADP + Pi + 2H2O → 2CO2 + 3NADH + FADH2 + ATP + 2H+ + CoASH
This must seem like a lot of work to produce just 1 ATP, especially since the partial, anaerobic oxidation of glucose in glycolysis produced in net fashion 2 ATPs. The key, however, is to realize that 3 NADHs and 1 FADH2 are produced, which when they are reoxidized in mitochondrial electron transport/oxidative phosphorylation, will produce multitudes of ATP.
As was true for glycolysis, this main energy-extracting pathway is highly regulated. We will this in the next section.
Figure \(\PageIndex{33}\) shows some key points about each reaction in the citric acid cycle.

Figure \(\PageIndex{33}\): Summary of the citric acid cycle.