
10.4: Working with Lipids
- Page ID
- 14979


Introduction
Lipids, although small compared to large biopolymers like proteins, nucleic acid, and large glycans, are very heterogenous in structure, given the large array of fatty acid and isoprenoid chain lengths, numbers of double bonds, etc that appear in different lipid classes. In addition to analyzing lipid structure, lipids are used in the laboratory to create liposomes (vesicles), which serve as models for membrane bilayers and encapsulation of chemical species (drugs, vaccines) for medical use and solubilization of membrane proteins. In this section, we will concentrate on the creation of lipid vesicles (critical for the encapsulation of RNA vaccines against the SARS CoV-2 spike protein) and the chemical analysis of biological lipids, whose composition affects health and disease states.
Liposomes
Liposomes produced in the lab can be unilamellar, consisting of a single bilayer surrounding the internal aqueous compartment, or multilamellar, consisting of multiple bilayers surrounding the enclosed aqueous solution. You can imagine the multilamellar vesicles resemble an onion with its multiple layers. Cartoons of unilamellar and multilamellar liposomes are shown in Figure \(\PageIndex{1}\), where each concentric circle represents a bilayer.
Liposomes vary in diameter. They can be generally categorized into small (S, diameter < 25 nm), intermediate (I, diameter around 100 nm), and large (L, diameter from 250-1000 nm). If these vesicles are unilamellar, they are abbreviated as SUV, IUV, and LUV, respectively. Their various sizes are shown in Table \(\PageIndex{1}\) below, in comparison to other large biological structures.
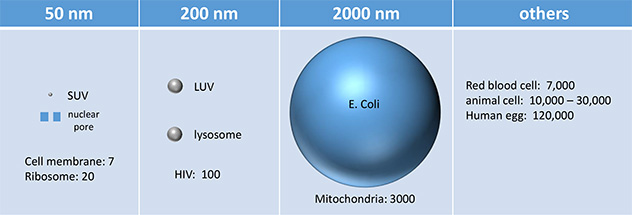
Table \(\PageIndex{1}\): Sizes of liposomes/vesicles compared to other biological structures
The chemical composition of liposomes can be widely varied. Most contain neutral phospholipids like phosphatidylcholine (PC), phosphatidyl ethanolamine (PE), or sphingomyelin (SM), supplemented, if desired, with negatively charged phospholipids, like phosphatidyl serine (PS) and phosphatidyl glycerol (PG). In addition, single-chain amphiphiles like cholesterol (C) and detergents can be incorporated into the bilayer membrane, which modulates the fluidity and transition temperature (Tm) of the bilayer. If present in too great a concentration, single-chain amphiphiles like detergents, which form micelles, can disrupt the membrane so completely that the double-chain amphiphiles become incorporated into detergent micelles, now called mixed micelles, in a process that effectively destroys the membrane bilayer.
Given the large degree of unsaturation at C2, what do you expect the transition temperature of a liposome composed only of egg yolk PC to be? (Vesicles made using more saturated PC from mammalian sources have Tm of around 40oC.) This high degree of unsaturation makes egg yolk PC very susceptible to oxidation, which could alter the properties of the liposome dramatically. Synthetic PC made with saturated fatty acids could alleviate that problem.
The properties of liposomes (charge density, membrane fluidity, and permeability) are determined by the lipid composition and size of the vesicle. The desired properties will be, in turn, determined by the use of the particular liposome. The vesicles offer wonderful, simple models to study the biochemistry and biophysics of natural membranes. Membrane proteins can be incorporated into the liposome bilayer using the exact method you will be using. But apart from these purposes, liposomes can be used to encapsulate water-soluble molecules such as nucleic acids, proteins, and toxic drugs. These liposomes can be targeted to specific cells if antibodies or other molecules which will bind specifically to the target cell can be incorporated into the bilayer of the vesicle. Intraliposomal material may then be transferred into the cell either by fusion of the vesicle with the cell or by endocytosis of the vesicle.
Since phospholipids will spontaneously form some type of bilayer structure when placed in water, most efforts in liposome production involve producing vesicles with the desired size, lamellar structure, and physical characteristics, which as previously stated are controlled both by liposome size and chemical composition. Also, ways must be developed to entrap the desired molecule inside the vesicle in the most cost-effective manner, and with minimal leaking of contents. All methods of production involve drying of organic solvent-solubilized lipids, dispersion of the lipids in the appropriate aqueous solution, and formation of monolamellar (one bilayer) liposomes or vesicles. Finally, the vesicles are characterized (chemical composition, Tm, permeability, size, etc.)
Drying of lipids
Purified lipids of the desired composition (often egg PC:cholesterol:PS in molar ratios of 0.9:1.0:0.1) are dissolved in a purified, water-free organic solvent mixture (often chloroform/methanol, 2:1 v/v) and dried down in a round bottom flask on a rotary evaporator under reduced pressure (using a water aspirator) and slightly elevated temperature (20-40oC). The rapid rotation of the flask will ensure that the lipid is dispersed over a large surface area, and will increase the rate of evaporation. To remove the last traces of solvent, the dried flask is usually placed under a high vacuum overnight. If a small volume (< 1 ml) of lipid solution is used, the solvent can be evaporated under a stream of nitrogen. To avoid entrapment of residual chloroform in the lipid film, the film is dissolved in t-butyl-methyl ether or diethylether, and dried several times. Alternatively, the residual solvent can be removed under a high vacuum.
Dispersion of the lipids
There are three main methods of dispersing lipids into an aqueous solution to form liposomes.
a. mechanical dispersion - in this method, lipid dried onto the inside glass surface of a container is hydrated with an aqueous solution, which peals off the lipid to form multilamellar - MLV - (multiple bilayers separated by water) vesicles. Only a small part of the aqueous solution is encapsulated inside the liposome, so this is not the method of choice for the encapsulation of expensive or rather insoluble solutes. Depending on the degree of agitation and the nature of the lipid used, different-sized liposomes can be prepared.
b. organic solvent dispersion - In these methods, the lipids, which are dissolved in organic solvents, are injected through a fine needle, at a slow rate, into an aqueous solution in which the organic solvent may be miscible (such as ethanol) or immiscible (such as ether). In each case, the lipids orient at the interface between the organic solvent and aqueous solution, to form bilayer structures. Injection of ethanol-dissolved lipids provides a simple way to produce SUV, but because liposome formation can not occur at an ethanol concentration greater than 7.5%, only a fraction of the total aqueous phase can be entrapped in the vesicle; hence this technique is not cost-effective for entrapment of an expensive solute. Alternatively, the lipid can be dissolved in ether and slowly injected into an aqueous solution which is warmed so that the ether evaporates at the rate at which it is injected. Since the ether is volatilized, large amounts of lipid can be introduced and the encapsulation efficiency of the aqueous solution is high.
c. detergent dispersion and solubilization - In this method, lipids are solubilized in an aqueous solution through the addition of detergents. The detergents are removed slowly from the solution, resulting in the spontaneous formation of liposomes. Detergents are single-chain amphiphiles that spontaneously form micelles in an aqueous solution when the concentration of free lipid rises to a minimum critical value, the critical micelle concentration (CMC); at this concentration, self-association of detergent results in the formation of a stable aggregate, the micelle. This is illustrated in Figure \(\PageIndex{2}\).

Table \(\PageIndex{2}\) below shows the properties and CMC of various detergents.
| Name | mM | mg/ml | MW |
|---|---|---|---|
| n-hepty glucopyranoside | 70 | 19.5 | 278 |
| n-octyl glucopyranoside | 23.2 | 6.8 | 292 |
| n-nonyl glucopyranoside | 6.5 | 2.0 | 306 |
| n-decyl maltoside | 2.19 | 1.1 | 499 |
| n-dodecyl maltotrioside | 0.2 | 0.16 | 825 |
| Triton X-100 (a) | 0.24 | 0.15 | 625 |
| Nonidet P-40 (b) | 0.29 | 0.02 | 603 |
| Tween 20 (c) | 0.033 | 0.04 | 1364 |
| Brij 98 (d) | 0.025 | 0.04 | 1527 |
| sodium deoxycholate | 2-6 | 1.7 | 415 |
| sodium taurocholate | 10-15 | 6.7 | 538 |
| sodium cholate | 14 | 6.0 | 431 |
| sodium dodecyl sulfate | 8.3 | 2.4 | 289 |
Making liposomes by dialysis
Lipids are deposited in a small container. An aqueous solution is then added, containing water-soluble molecules for encapsulation. Detergent is then added at a concentration in excess of the lipid concentration and greater than its CMC. The lipid molecules are then "emulsified" in the detergent micelle. The solubilized mixture is then placed in a semi-permeable dialysis bag, which is placed in a large volume of an aqueous solution. The free detergent in solution is in equilibrium with the detergent in the micelle. The bag contains microscopic holes large enough for the monomeric detergent molecule to pass through, but small enough so that the large micelle can not. The lipid, during this process, is embedded in the micelle forming a detergent-lipid mixed micelle. As dialysis continues, the monomeric detergent partitions throughout both the volume in the bag and the volume surrounding the bag, while the mixed micelle remains in the bag. If the aqueous solution surrounding the bag is changed several times with fresh solution, the equilibrium in the bag is shifted to the monomeric form. Alternatively, detergent-adsorbing beads (such as Bio Bead SM-2 by Bio-Rad) can be placed in the aqueous solution surrounding the bag to speed up the process of detergent re-equilibration. Eventually, all the detergent is in this form, and during the process, which occurs slowly, the lipid in the mixed micelle self-associates to form a liposome. A detergent of low monomer molecular weight and a high CMC is most desirable for this method of liposome production. Another method of removing the free detergent is through gel filtration chromatography. In this technique, molecules of disparate molecular weights can be separated from each other. An explanation follows this discussion. This method of formation of unilamellar liposomes is the method of choice if membrane proteins are to be inserted into the liposome bilayer to target the liposome. It is not the best method, however, for quantitative encapsulation of expensive soluble molecules.
Making Liposomes by Extrusion
These multilamellar liposomes can be further processed to form unilamellar liposomes by several techniques. These include probe or bath sonication of the MLVs, extrusion at high pressure of the MLVs through membrane filters of defined pore size, or pH-induced vesiculation in which a transient change in pH destabilizes the MLVs in favor of unilamellar liposomes. Another technique involves the fusion of SUVs by repeated freezing and thawing or by fusion of SUVs containing acidic phospholipids (such as PS) through Ca2+-mediated aggregation.
Figure \(\PageIndex{3}\): below shows the structure of vesicles as they undergo multiple freeze/thaw cycles.

Figure \(\PageIndex{4}\) shows the final step in making large unilamellar vesicles by extrusion of freeze/thaw intermediates.

Once the liposomes are formed, they must be separated from free monomeric lipids, detergents, and unencapsulated solutes. This can be done again by dialysis, or more readily by size exclusion chromatography. Macromolecules of different sizes can be separated on a column in which the stationary phase is a polymerized agarose or acrylamide bead, which contains pores of various sizes. A small molecule (such as monomer detergent, free lipid, or small aqueous solute) in the mobile phase (aqueous buffered solution) may enter the pores in the bead, while a larger macromolecule or aggregate (such as a large protein, a micelle, or a liposome) may not, due to size restriction. The result is that a larger fraction of the overall volume of the column is available to the smaller molecules, which thus spend a longer time on the column and are eluted by the mobile solvent after the larger species. Liposomes can be characterized both chemically, to determine the average lipid and protein (if they were incorporated) composition of the bilayer, and physically, to determine the size, permeability, lamellarity, and amount of encapsulated material. Size is usually determined by electron microscopy or indirectly by light scattering from these large species.
Chemical Analysis of Lipids
There are so many lipids and their derivatives that are so subtlety different that the most sensitive ones are needed for separation and analysis. The field of lipidomics focuses on the analysis of the structure and function of all lipids in the cell. The most useful techniques are analysis (and separation) by gas chromatography (GC) followed by mass spectrometry (MS). NMR spectroscopy is also important. For GC analysis, the lipid must be made volatile, which limits its use in some circumstances. MS analysis is the most sensitive. MS requires ion formation, and techniques like electrospray ionization and matrix-assisted laser desorption/ionization (MALDI) are used.
Gas chromatography
(This section is adapted from https://www.intechopen.com/chapters/64008. Creative Commons Attribution 3.0 License). In GC, gas is the mobile phase that carries lipids through the stationary phase of the column. Gas chromatography (GC) is used to separate organic compounds from a mixture in the gas form. For this purpose, the GC uses interactions among the sample components and the stationary/mobile phases. After lipid extraction with chloroform and/or methanol, the samples (lipid mixture) are usually liquids and must be exposed to a high temperature at the gas chromatograph entrance (injector). Vaporized, the samples are carried by a gas, which is usually a nonheavy and inert gas (i.e., hydrogen, helium), through a long capillary column containing a high or low polarity material (stationary phase)
The gaseous compounds generated from the vaporized sample interact with the stationary phase which allows each compound to elute/separate at a different time (retention time). Because GC considers both the chemical and physical properties of the vaporized compounds, those with more chemical affinity to the stationary phase will take a longer time to be removed from the column and the temperature will influence the overall process. This explains why the column stays in an oven, which is programmed to work at different temperature ranges (i.e., temperature programming) in which the compounds are carried out by the gas according to their boiling point until they get to an electronic detector.
At the end of GC analysis, the electronic detector generates a chromatogram based on retention time by intensity. This allows a qualitative identification of the lipid compounds by comparing their retention times with certified standards using the flame ionization detector (FID) or by deduction of spectra information using a mass spectrometer as a detector. Lipid quantification can also be performed using analytical procedures of external or internal certified standards in GC analysis.
The main points to be considered when assessing FAs by GC analysis are the carrier gas flow rate, column length, and temperature because these can influence the order or retention time of the lipid compounds and then must be precisely standardized. The column length of the stationary phase influences the resolution of the analytes, once the number of theoretical plates (hypothetical zone in which two phases establish an equilibrium with each other) is respectively high in the longer column. As fat and oils have high boiling points not supported by the stationary phase, a previous derivatization reaction step is required after lipid extraction from the biological sample, in which triacylglycerol and free fatty acids are transformed into their respective free fatty esters with lower boiling points (transesterification/esterification reaction). Several methods are available for FAs derivatization.
Particularly for cholesterol analysis, the sample preparation must consider a derivatization reaction. This allows to block protic sites of steroids obtained after an unsaponifiable lipid extraction had been performed, and also, diminishes dipole-dipole interactions, increases the volatility of the compounds, and to generate products with reduced polarity. Cholesterol derivatization is usually achieved by using trimethylsilyl compounds as reagents (silylation reaction). A common method for this purpose uses N,O-bis(trimethylsilyl-trifluoroacetamide/trimethylchlorosilane).
Mass spectrometry
(This section is derived from https://doi.org/10.1038/s41467-019-14180-4. Creative Commons Attribution 4.0 International License: http://creativecommons.org/licenses/by/4.0/).
Mass spectrometry (MS) has become the method of choice for lipid analysis, offering label-free detection at high sensitivity and structural characterization capability. However, large-scale lipid analysis with a comprehensive capability of revealing all levels of structure information still represents a significant analytical challenge for lipidomics. General protocols, for instance, have five levels in terms of structure information, including lipid class, fatty acyl identities, fatty acyl sn-positions, and C=C location/geometry (viz cis/trans) in the fatty acyl. Successful attempts for determining C=C locations in fatty acyls or their sn-positions have already been reported for MS analysis, enabling the characterization of detailed structure moieties and identification of lipid structure isomers. An extremely useful feature offered by lipid isomer analysis is the relative quantitation achieved at high precisions without requiring the use of lipid standards, which are not readily available. Remarkably, our recent study demonstrated a close correlation between the lipid C=C location isomer compositions and Type II diabetes, which owes to tighter regulation on lipid desaturation, allowing efficient elimination of interferences due to variations among samples"
Various methods have been explored for differentiating the lipid C=C location and sn-position isomers. Ozone-induced dissociation (OzID) and ultraviolet photodissociation (UVPD) have been used to determine both sn-positions and C=C locations in GPs. By coupling the Paternò-Bǜchi (PB) photochemical reaction with tandem MS (MS/MS), qualitative and quantitative analysis of lipids with C=C specificity from complex biological samples can be accomplished. PB reaction converts the C=C to an oxetane which can be preferentially fragmented by low-energy collision-induced dissociation (CID).
An ideal analytical tool for lipidomics to survey a wide range of lipids in discovery work should not only provide detailed information at multiple lipid structure isomer levels (e.g., C=C location/geometry and sn-position), but also be feasible for large-scale quantitative analysis. UVPD is capable of assigning C=C locations and sn-positions of fatty acyls, while OzID may be the only one that has been well demonstrated for assigning C=C locations in sn-specific fatty acyls. One problem of OzID is the long reaction time required for the ion trap implementation, however recent work has demonstrated the ability to perform OzID on LC-compatible time scales in the high-pressure regions of the MS system. For the PB reaction method, both shotgun analysis and HPLC-PB-MS/MS workflow have been developed for identifying a large number of C=C location isomers. Figure \(\PageIndex{5}\) describes methods for the determination of C=C location and sn-isomers in lipids.
