
9.1: DNA Isolation, Sequencing, and Synthesis
- Page ID
- 14969


Genomic and complementary DNA
The ability to sequence the DNA of an organism has revolutionized our understanding of biology and evolution. DNA can be isolated directly from living, dead, and even extinct species and the "primary" sequence of A, G, C, and T bases in the molecule determine. We can read (sequence), write (synthesize), and edit (mutate) DNA at will. Before we explore how to purify, sequence (read) and synthesize (write) DNA, it's important to differentiate two different types of DNAs, genomic and complementary DNA (cDNA), which is made by reverse transcribing messenger RNA into DNA. Since mRNA has no nucleotides encoded by introns, c-DNA provides just the coding sequences for protein.
Genomic deoxyribonucleic acid (gDNA) is chromosomal DNA, which does not include the extra-chromosomal DNA found in the mitochondria of eukaryotes or plasmids in bacteria (plasmids will be discussed in more detail in section 5.3 during the discussion of gene cloning and expression). Most organisms have the same genomic DNA in every cell (one exception is the genomic DNA for antibodies in B cells and T cells receptors in T-cells that have been altered as the cells become more terminally differentiated. It is also important to remember that only certain genes are active (expressed) in each cell. The subset of expressed genes is specific to a given differentiated cell and allows for the expression of specific cell functions. Liver cells, for example, don't express the gene for the protein opsin, which is expressed in retinal cells and is required for vision.
The genome of an organism (encoded by the genomic DNA) is the (biological) information of heredity that is passed from one generation of organism to the next. That genome is transcribed to produce various RNAs, which are necessary for the function of the organism. Precursor mRNA (pre-mRNA) is transcribed by RNA polymerase II in the nucleus. pre-mRNA is then processed by splicing to remove introns, leaving the exons in the mature messenger RNA (mRNA). Additional processing includes the addition of a 5' cap and a poly(A) tail to the pre-mRNA. The mature mRNA may then be transported to the cytosol and translated by the ribosome into a protein. Other types of RNA include ribosomal RNA (rRNA) and transfer RNA (tRNA). These types are transcribed by RNA polymerase II and RNA polymerase III, respectively, and are essential for protein synthesis. However, 5s rRNA is the only rRNA that is transcribed by RNA Polymerase III. cDNA is derived from mRNA, so it contains only exons but no introns.
Figure \(\PageIndex{1}\) shows the flow of information stored in eukaryotic DNA and its eventual expression in mRNA.

Red indicates coding exons, which are separated by gray introns. At the beginning and end of a gene sequence (encoded by 3 exons in the figure below) are 5' and 3' untranslated regions (UTRs) which are also transcribed and are represented in the mature mRNA. Also, there are potential regulatory sequences, which are not transcribed (yellow) on both sides of the transcribable part of the gene. The 5'-end promoter is where transcription factors and RNA polymerase assemble before the start of transcriptions. In addition, there are regulatory enhancers and silencers more distal from the gene sequences. An open reading frame (ORF) is a region of the DNA that can be decoded into a mRNA and doesn't have a stop signal (codon) in it that would prematurely terminate transcription.
In contrast, complementary DNA (cDNA) is synthesized from a single-stranded RNA (e.g., messenger RNA (mRNA) or microRNA) template in a reaction catalyzed by the enzyme reverse transcriptase. Reverse transcriptase is an enzyme found in retroviruses such as HIV that have RNA as their core genetic material. Upon entering the host cell the RNA is reverse-transcribed to produce a copy of cDNA that can then integrate into the host's genomic DNA. In biotechnology, reverse transcriptase is often used to create cDNA from the mRNA expressed in specific cells or tissues. In this way, the eukaryotic genes can be cloned without any introns housed in the structure. This is especially useful if the goal is to express the protein in a prokaryotic (bacterial) host. Recall that bacterial DNA does not contain any intron sequences within its chromosomal DNA. Thus, if you are using a prokaryotic system to express eukaryotic proteins, you must use cDNA, as the prokaryotic system will not be able to remove intron sequences following gene transcription. The term cDNA is also used, typically in a bioinformatics context, to refer to an mRNA transcript's sequence expressed as DNA bases (GCAT) rather than RNA bases (GCAU).
The gene organization of prokaryotes is different in that they don't have introns. In addition, some genes for a common pathway for example are continuous in the DNA. These stretches of DNA are called operons. Transcription from an operon produces a polycistronic RNA transcript. The words cis and are used in chemistry to describe R groups on the same size (cis) or opposite sides (trans) of a double bond. In DNA, cis-elements are in a single DNA section while trans-elements usually refer to proteins (away from the gene) binding to the DNA. Hence the term polycistronic for bacterial operons (with multiple genes sequentially arranged in the DNA sequence). Figure \(\PageIndex{2}\) shows the organization of prokaryotic gene structure.

DNA Extraction/Purification
The first isolation of DNA was done in 1869 by Friedrich Miescher. Now purification kits are available from multiple manufacturers.
DNA can be isolated from whole tissue or cell cultures. Let's consider just DNA extraction from cells grown in the lab. Cells are first collected by centrifugation and then treated with detergents like sodium dodecyl sulfate to lyse the cell membranes. Proteases and DNAase-free RNAase can be added to digest proteins and RNA, respectively.
Methods involving phenol/chloroform extractions:
In older methods, a mixture of phenol and chloroform or phenol/chloroform/isoamyl alcohol is used to extract DNA from the solution. Students who have performed liquid/liquid extractions in chemistry lab courses should recognize that this will form a biphasic mixture with water. Nonpolar substances like lipids and cellular debris will partition into the nonpolar phase (chloroform/phenol) or in the interface between them (as suspended insoluble material). Chloroform is very dense as it contains a chlorine atom). Phenol is somewhat soluble in water (8 g/100 g water) but very soluble in chloroform. In mixing during the extraction, the dissolved phenol alters the properties of water sufficiently enough to push the delicate equilibrium of proteins from the native to the denatured states, which aggregate and precipitate. On settling, DNA will remain in the aqueous phase. The use of chloroform/phenol in DNA extractions has a potential problem. Phenol (hydroxylated benzene) can lose one electron from the oxygen atom forming a free radical, which can be stabilized by resonance with pi electrons in the aromatic ring. Free radicals can damage DNA, so most new methods of purification do not use phenol/chloroform extractions.
Most methods involve precipitating the extracted DNA at some point in the purification process using cold ice-cold ethanol or isopropanol. DNA is to a first approximation a long polyanion so it would be very difficult to purify "naked" DNA from solution since the extensive negative charges on the DNA would prevent aggregate and precipitate formation. This is not a problem if the ionic strength of the medium is sufficiently high so bound positively charged counter-ions can shield the negative charges from each other, allowing precipitation.
Methods involving adsorption chromatography using silica gel: Nucleic acids bind or adsorb to a solid phase (silica or other) depending on the pH and the salt concentration of the buffer. If small amounts are needed (such as for isolating recombinant plasmid from bacteria), small spin columns are employed. This method relies on the fact that nucleic acid will bind to the solid phase of silica gel under certain conditions and then be released when those conditions are altered. These features are illustrated in Figure \(\PageIndex{3}\).
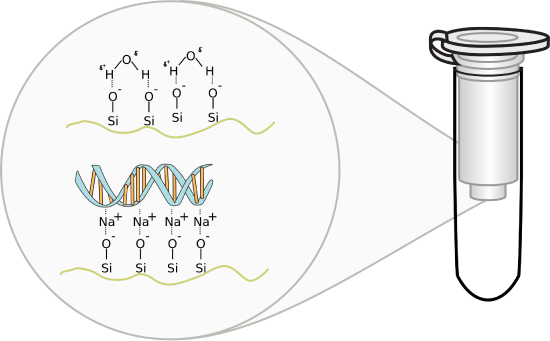
Figure \(\PageIndex{3}\): DNA-Silica gel interactions. Image by Squidonius
The solution containing DNA is applied to a small spin column containing silica gel and placed in a mini-centrifuge. On spinning, the nucleic acid will bind to the silica gel membrane as the solution passes through. After multiple "spin" washes to remove non-specific cellular components from the column, the DNA is eluted with a low salt elution buffer (or simply water). Unlike RNA which degrades very quickly, DNA is quite stable and can be stored for long periods at -20oC.
Many student readers have used silica gel chromatography or silica gel thin-layer chromatography to separate and analyze organic mixtures. These techniques are usually performed in a mixture of organic solvents (like hexane/ethyl acetate for example). The use of silica gel to purify DNA from an aqueous solution might seem strange so we will briefly explore how DNA binds.
In silica gel, each silicon atom is tetrahedrally bonded (sp3 hybridization) to four oxygen atoms, with each oxygen atom covalently linked to two silicon atoms. At the surface of the particle, the oxygen atoms are capped with H atoms, so the entire surface contains a "sea" of OHs, and therefore hydrogen bond donors and acceptors. At lower pHs, some could ionize to form O- ions. If salt concentrations are high enough, the percentage of O- ions increases, since they are stabilized by the cations in the salt (shifting the equilibrium to the ionized state).
DNA, a long negatively charged anion, can bind to the silica surface using two types of noncovalent interaction. It can form hydrogen bonds with the silica gel surface OHs. In addition, it can interact with the surface through ion-ion interactions mediated by bridging cations (like Na+ from the high-concentration salt solution) as illustrated in the figure above. The binding solutions used in the spin column adsorption steps have high concentrations of chaotropic salts that disrupt water structure and hydrogen bonding. The salts also denature proteins and in effect dehydrate the DNA. Some chaotropic salts used include sodium iodide, sodium perchlorate, guanidinium thiocyanate, and guanidinium chloride. The sodium or guanidinium acts as bridging cations, allowing the adsorption of the negative DNA to negative charges on the silica gel surface. Sodium acetate and Tris-HCl are included to buffer pH from 6-7. Now it becomes easy to understand how pure water or low salt concentration solutions elute the bound DNA after extensive column washing since pure water or low salt solutions would strip the bound intermediary cations from the silica column.
After isolation, the DNA is dissolved in a slightly alkaline buffer, usually in a Tris-EDTA buffer, or in ultra-pure water. EDTA binds divalent cations like Ca2+, which activate nucleases. Modifications made to these standard techniques are often done if the tissue being used is difficult to break down, if contaminants persist in the lysis solution that inhibit further reactions, or if the sample is extremely minimal, as is often the case in forensic investigations. In addition, different commercial kits will be tailored for the isolation of larger genomic DNA or smaller plasmid DNA.
The purity of a DNA preparation is usually determined by measuring the absorbance of the solution at 230, 260 (peak absorbance nucleic acids), and 280 nm (peak absorbance proteins), often using an instrument that requires a tiny droplet of solution. Figure (\PageIndex{4}\) below shows the relative absorbance spectra of proteins and nucleic acids.
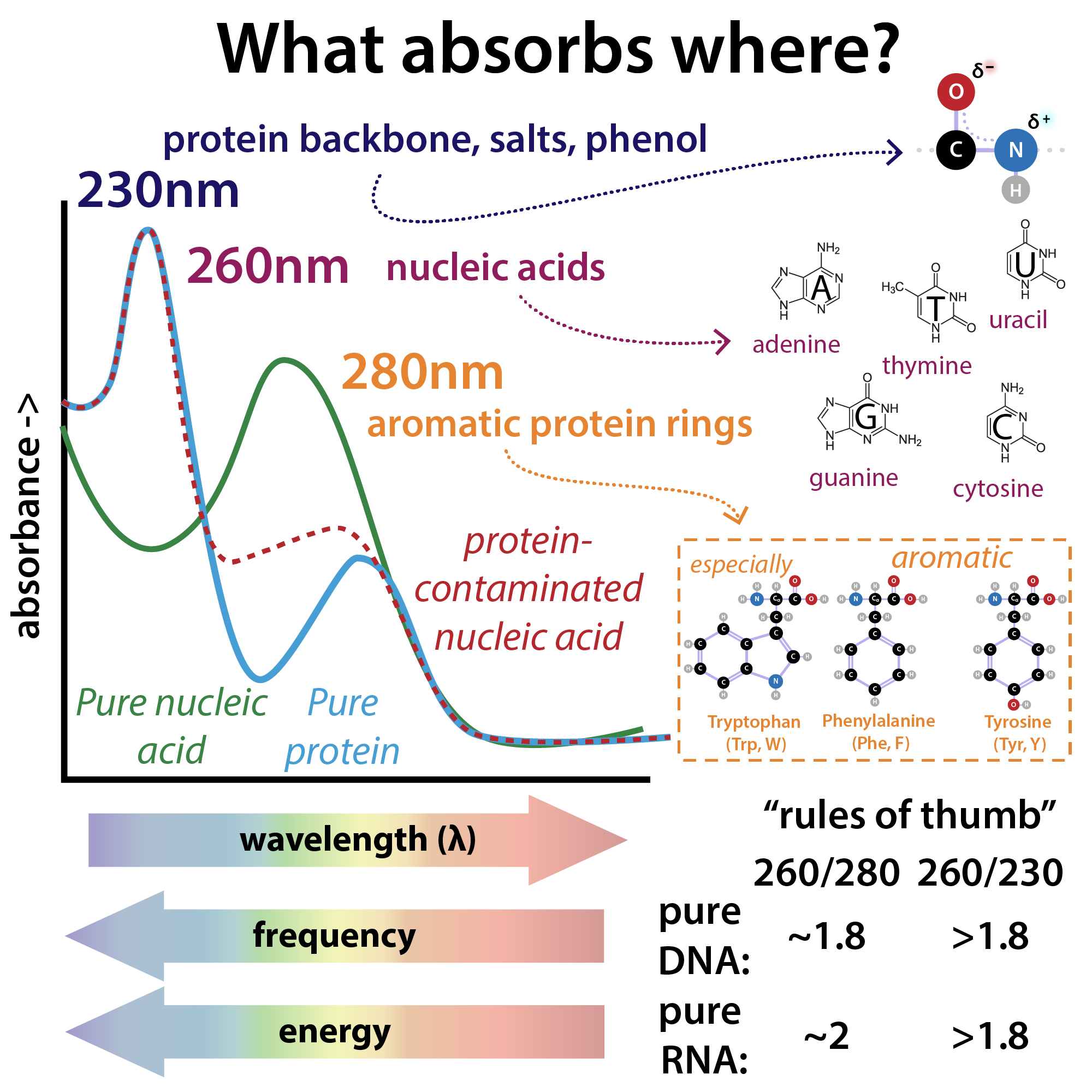
Figure (\PageIndex{4}\) relative absorbance spectra of proteins and nucleic acids. Brianna Bibel. The Bumbling Chemist https://thebumblingbiochemist.com/36...purity-ratios/
The A260/A280 gives a measure of protein contamination, with a value around 1.8 indicating "pure" DNA. The A260/A230 gives information on protein and other solution contamination. A pure solution of DNA that has an A260 = 1.0 has a concentration of 50 μg/mL = 50 ng/μL.
When the temperature of a dsDNA solution is increased, in some range of temperature the A260 increases by about 37%. This is called the hyperchromic effect which occurs when the bases in DNA unstack on denaturation of dsDNA to single-stranded DNA as the intrastrand hydrogen bonds break. Figure (\PageIndex{5}\) below shows a graph of A260 vs temperature. The midpoint of the cooperative change in the absorbance signifies that the population of DNA molecules is 50% denatured.

Given the cooperative of unfolding, it is less likely that the population consists of individual molecules that are 50% denatured (i.e. a single molecule being 50% double-stranded and 50% single-stranded). Accordingly, a lower concentration (33 μg/mL) of fully single-stranded DNA gives an A260 = 1.
DNA Sequencing Techniques
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Knowledge of DNA sequences has become indispensable for basic biological research and in numerous applied fields such as medical diagnosis, biotechnology, forensic biology, virology, and biological systematics. Comparing healthy and mutated DNA sequences can diagnose different diseases including various cancers, characterize antibody repertoire, and can be used to guide patient treatment. Having a quick way to sequence DNA allows for faster and more individualized medical care to be administered, and for more organisms to be identified and cataloged.
The rapid speed of sequencing attained with modern DNA sequencing technology has been instrumental in the sequencing of complete DNA sequences, or genomes, of numerous types and species of life, including the human genome and other complete DNA sequences of many animal, plant, and microbial species.
The first DNA sequences were obtained in the early 1970s by academic researchers using laborious methods based on two-dimensional chromatography. Following the development of fluorescence-based sequencing methods with a DNA sequencer, DNA sequencing has become easier and orders of magnitude faster.
The canonical structure of DNA has four bases: thymine (T), adenine (A), cytosine (C), and guanine (G). DNA sequencing is the determination of the physical order of these bases in a molecule of DNA. However, DNA bases are often modified by epigenetic processes to control gene expression. Thus, many other modified bases may be present in a DNA molecule than the standard four bases. For example, in some viruses (specifically, bacteriophages), cytosine may be replaced by hydroxymethyl- or hydroxymethylglucose cytosine. In eukaryotic DNA, variant bases with methyl groups or phosphosulfate may be found as shown in Figure (\PageIndex{6}\) below. Depending on the sequencing technique, a particular modification, e.g., the 5mC (5 -methylcytosine) common in humans, may or may not be detected.

Early DNA sequencing methods
The first method for determining DNA sequences involved a location-specific primer extension strategy established by Ray Wu at Cornell University in 1970. DNA polymerase catalysis and specific nucleotide labeling, both of which figure prominently in current sequencing schemes, were used to sequence the cohesive ends of lambda phage DNA. Between 1970 and 1973, Wu, R Padmanabhan, and colleagues demonstrated that this method can be employed to determine any DNA sequence using synthetic location-specific primers. Frederick Sanger then adopted this primer-extension strategy to develop more rapid DNA sequencing methods at the MRC Centre, Cambridge, UK, and published a method for "DNA sequencing with chain-terminating inhibitors" in 1977. Walter Gilbert and Allan Maxam at Harvard also developed sequencing methods, including one for "DNA sequencing by chemical degradation". In 1973, Gilbert and Maxam reported the sequence of 24 base pairs using a method known as wandering-spot analysis. Advancements in sequencing were aided by the concurrent development of recombinant DNA technology, allowing DNA samples to be isolated from sources other than viruses.
Maxam-Gilbert sequencing requires radioactive labeling at one 5' end of the DNA and purification of the DNA fragment to be sequenced. Chemical treatment then generates breaks at a small proportion of one or two of the four nucleotide bases in each of the four reactions (G, A+G, C, C+T). The concentration of the modifying chemicals is controlled to introduce on average one modification per DNA molecule. Thus a series of labeled fragments is generated, from the radiolabeled end to the first "cut" site in each molecule. The fragments in the four reactions are electrophoresed side by side in denaturing acrylamide gels for size separation. To visualize the fragments, the gel is exposed to X-ray film for autoradiography, yielding a series of dark bands each corresponding to a radiolabeled DNA fragment, from which the sequence may be inferred.
The technical aspects of Maxam-Gilbert sequencing caused it to go out of favor once the Sanger sequencing method had been well established, as described below.
Sanger Sequencing Method
The chain-termination method developed by Frederick Sanger and coworkers in 1977 soon became the method of choice, owing to its relative ease and reliability. When invented, the chain-terminator method used fewer toxic chemicals and lower amounts of radioactivity than the Maxam-Gilbert method. Because of its comparative ease, the Sanger method was soon automated and was the method used in the first generation of DNA sequencers.
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotide triphosphates (dNTPs), and modified di-deoxynucleotide triphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3'-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease the extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labeled for detection in automated sequencing machines.
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP, and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones as shown in (\PageIndex{7}\) below.

The dideoxynucleotide concentration should be approximately 100-fold lower than that of the corresponding deoxynucleotide (e.g. 0.005mM ddTTP : 0.5mM dTTP) to allow enough fragments to be produced while still transcribing the complete sequence. In total, four separate reactions are needed in this process to test all four ddNTPs. This is illustrated in Figure (\PageIndex{8}\) below.
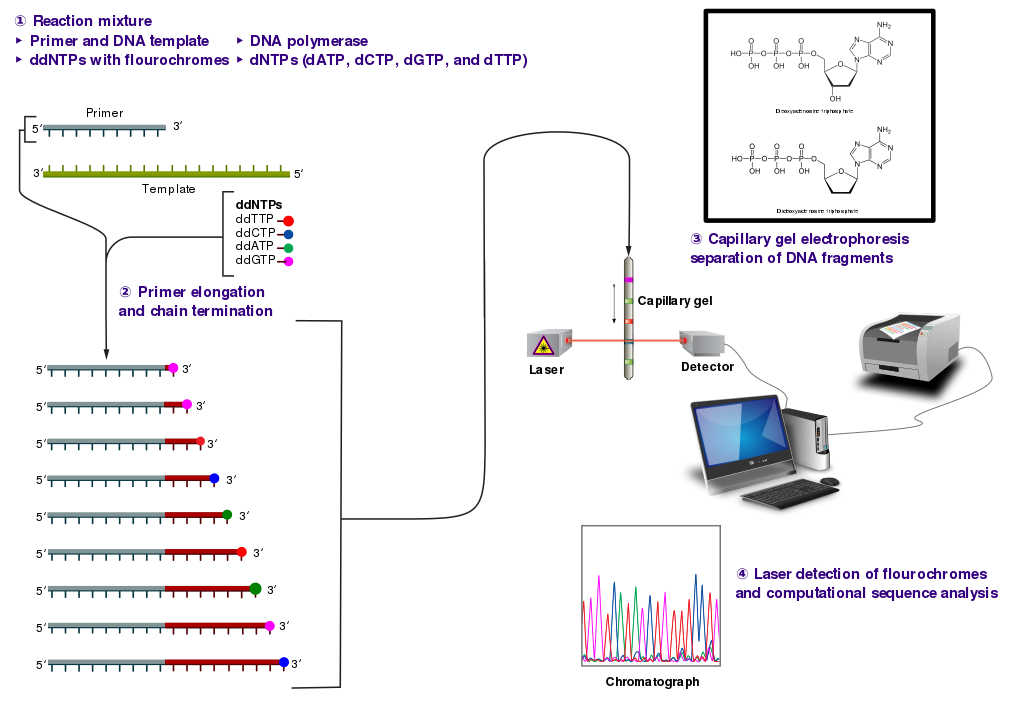
Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. This technique was frequently performed using a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C). The DNA bands may then be visualized by autoradiography or UV light and the DNA sequence can be directly read off the X-ray film or gel image, as shown in Figure (\PageIndex{9}\) below.
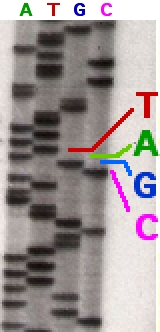
Figure (\PageIndex{9}\): Traditional Sanger Sequencing Gel. Sequence visualized by autoradiography. Each lane contains a single reaction that has all four regular nucleotides and a small amount of one of the dideoxynucleotides (ddNTPs). Over time, the ddNTPs will be incorporated at each position containing that specific nucleotide. The gel can then be read from the bottom to the top, as the smallest fragments (those fragments terminated the closest to the primer at the 5'-end) will run the farthest distance in the gel. The sequence of this fragment is 5'-TACGAGATATATGGCGTTAATACGATATATTGGAACTTCTATTGC-3'. Image by John Schmidt
Automation of the Sanger sequencing method was made possible when the shift from radioactively tagged nucleotides to fluorescently tagged nucleotides was made. Within the automated sequencers, capillary gel electrophoresis is performed rather than separating the samples using gel electrophoresis. The output from capillary electrophoresis are fluorescent peak trace chromatograms, as shown in Figure (\PageIndex{10}\) below. Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch. Batch runs may occur up to 24 times a day greatly enhancing the speed with which samples may be sequenced and analyzed. Common challenges of DNA sequencing with the Sanger method include poor quality in the first 15-40 bases of the sequence due to primer binding and deteriorating quality of sequencing traces after 400-500 bases.

Figure (\PageIndex{10}\): Side by Side Comparison of Gel Electrophoresis and Capillary Electrophoresis. Lefthand Diagram shows the traditional autoradiogram of Sanger sequencing samples. The Righthand Diagram shows the same reactions using fluorescently tagged ddNTPs separated by capillary electrophoresis. The chromatogram output is shown on the far right. Image by Abizar
Sanger sequencing is the method that prevailed from the 1980s until ~2005. Over that period, great advances were made in the technique, such as fluorescent labeling, capillary electrophoresis, and general automation. These developments allowed much more efficient sequencing, leading to lower costs. The Sanger method, in mass production form, is the technology that produced the first human genome in 2001, ushering in the age of genomics.
Microfluidic Sanger Sequencing
Microfluidic Sanger sequencing is a lab-on-a-chip application for DNA sequencing, in which the Sanger sequencing steps (thermal cycling, sample purification, and capillary electrophoresis) are integrated on a wafer-scale chip using nanoliter-scale sample volumes (Figure (\PageIndex{11}\)). This technology generates long and accurate sequence reads while obviating many of the significant shortcomings of the conventional Sanger method (e.g. high consumption of expensive reagents, reliance on expensive equipment, personnel-intensive manipulations, etc.) by integrating and automating the Sanger sequencing steps.
.jpg?revision=1&size=bestfit&width=370&height=260)
Next-generation sequencing (NGS)
Next-generation sequencing (NGS), also known as high-throughput sequencing, is the catch-all term used to describe several different modern sequencing technologies. These technologies allow for the sequencing of DNA and RNA much more quickly and cheaply than the previously used Sanger sequencing, and as such revolutionized the study of genomics and molecular biology. We present information from specific companies that have developed these new technologies without endorsements. Such technologies include:
Illumina Sequencing - In NGS, vast numbers of short reads are sequenced in a single stroke using the lab-on-a-chip technology described above. To do this, the input sample must be cleaved into short sections. In Illumina sequencing, 100-150bp reads are used. Somewhat longer fragments are ligated to generic adaptors and annealed to a slide using the adaptors. PCR is carried out to amplify each read, creating a spot with many copies of the same read. They are then separated into single-stranded DNA to be sequenced as shown in Figure (\PageIndex{12}\) below.

Roche 454-Sequencing is similar to the Illumina process but can sequence much longer reads. Like Illumina, it does this by sequencing multiple reads at once by reading optical signals as bases are added. As in Illumina, the DNA or RNA is fragmented into shorter reads, in this case, up to 1kb (1,000bp). Generic adaptors are added to the ends and these are annealed to beads, one DNA fragment per bead. The fragments are then amplified by PCR using adaptor-specific primers. Each bead is then placed in a single well of a slide with each well containing a single bead, covered in many PCR copies of a single sequence. The wells also contain DNA polymerase and sequencing buffers (Figure (\PageIndex{13}\).

Newer technologies such as the Ion Torrent Technology detect sequence data using electrical signals on a semiconductor chip, rather than optically reading dye-labeled nucleotides. This is possible as the addition of a dNTP to the DNA polymer causes the release of an H+ ion (Figure (\PageIndex{14}\)). As in other kinds of NGS, the input DNA or RNA is fragmented, this time ~200bp. Adaptors are added and one molecule is placed onto a bead. The molecules are amplified on the bead by emulsion PCR. Each bead is placed into a single well of a slide. This is illustrated in Figure (\PageIndex{14}\) below.

Nanopore Sequencing
In this technique, flow cells are constructed which contain nanopores in a nonlipid membrane that is resistant to current flow. When a voltage is applied across a membrane separating two salt solutions, the current through each nanopore channel can be detected by a sensor chip. When a larger molecule moves through the pore, a disruption in basal current occurs. Computer algorithms have been developed that detect base-specific changes in the current as the base (even chemical-modified ones) moves through the membrane. The sequence is then decoded in real-time.
Single-stranded DNA or RNA can be driven through the pore by a transmembrane potential in a process similar to electrophoresis. The enzyme DNA helicase, a motor protein, can be attached to the outer part of the pore protein. This enzyme binds single-stranded DNA and moves along the DNA in a process that requires ATP. If the helicase is attached to the pore protein, the single DNA would then move, allowing control of its movement through the pore.
The nanopores are made of real membrane proteins (which we discuss in Chapter 11). One example is α-hemolysin, a heptamer that has an inner pore diameter of 1 nm. When embedded in real cells, it can allow the flow of K+ (diameter around 250 pm = 0.25 nm) and other ions across the cell membrane, changing the osmotic balance and lysing the cell. The pore size of the proteins used in nanopore sequencing allows single-stranded DNA to flow through it. An interactive iCn3D model of protein used for nanopore sequencing, Curli transport lipoprotein CsgG (4uv3), is shown in the membrane bilayer in Figure \(\PageIndex{15}\).
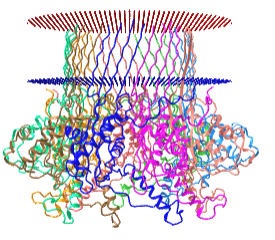_for_DNA_nanapore_sequencing.png?revision=1)
 Figure \(\PageIndex{15}\): Curli transport lipoprotein CsgG (4uv3) for DNA nanopore sequencing. (Copyright; author via source).
Figure \(\PageIndex{15}\): Curli transport lipoprotein CsgG (4uv3) for DNA nanopore sequencing. (Copyright; author via source).
Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...LC5K3vSjYjtHN7
Nanopore technologies have enabled the production of small hand-held DNA sequencing devices that can be plugged into the USB drive on a laptop computer and utilized in the field under real-time collection conditions. Future modifications might replace protein pores with synthetic solid-state nanopores. For instance, the membrane might be made of graphene with pores of a specific size made in it.
Figure \(\PageIndex{16}\) shows an animation of a single-stranded DNA moving through a protein pore (blue) assisted by a motor protein (magenta)

Single Molecule, Real-Time (SMRT) sequencing
In this technique, either RNA or DNA is converted to dsDNA. Deoxynucleotide "adapters" are added to connect the 5'-end of strand 1 to the 3'-end of strand 2 and another adapter to connect the 3'-end of strand 1 to the 5'-end of strand 2 resulting in a "circular" ss-DNA molecule. This single molecule is then drawn into a nanophotonic nanowell made in a thin metal film deposited on glass. The dimensions of each well allow only a single circular ss-DNA molecule. Hundred of different circular ss-DNA molecules entering individual wells are shown in Figure \(\PageIndex{17}\). The blue stretches represent the adapters.

The wells are approximately 100 nm in diameter. DNA polymerase and dNTPs can be added to the nanowells, which contained a single molecule of immobilized circular ssDNA. The DNA is immobilized by its biotinylated or attachment to magnetic beads which interact with streptavidin-coated wells. When confined to the wells the apparent concentration of reactants for the polymerization can be quite high, allowing robust DNA polymerase activity.
DNA sequencing using real-time fluorescence monitoring can be done in a massively parallel fashion. The fluorophore is connected to the terminal phosphate of the dNTP. When a phosphodiester bond is made by the DNA polymerase, the fluorophore is released as a leaving group, and the result is a natural, unmodified DNA that continues to grow. The YouTube video below shows the entire process of a single molecule, real-time sequencing.
The four main advantages of Next Generation Sequencing (NGS) over classical sequencing are described below.
Sample size
NGS is significantly cheaper, quicker, needs significantly less DNA, and is more accurate and reliable than Sanger sequencing. Let us look at this more closely. For Sanger sequencing, a large amount of template DNA is needed for each read. Several strands of template DNA are needed for each base being sequenced (i.e. for a 100bp sequence you'd need many hundreds of copies, for a 1000 bp sequence you'd need many thousands of copies), as a strand that terminates on each base is needed to construct a full sequence. In NGS, a sequence can be obtained from a single strand. In both kinds of sequencing multiple staggered copies are taken for contig construction and sequence validation.
Speed
NGS is quicker than Sanger sequencing in two ways. Firstly, the chemical reaction may be combined with the signal detection in some versions of NGS, whereas in Sanger sequencing these are two separate processes. Secondly and more significantly, only one read (maximum ~1kb) can be taken at a time in Sanger sequencing, whereas NGS is massively parallel, allowing 300Gb of DNA to be read on a single run on a single chip.
Cost
The reduced time, people power, and reagents in NGS mean that the costs are much lower. The first human genome sequence cost in the region of $2.7 billion in 2003. Using modern Sanger sequencing methods, aided by data from the known sequence, a full human genome still cost $300,000 in 2006. Sequencing a human genome with NGS today costs roughly $1,000.
Accuracy
Repeats are intrinsic to NGS, as each read is amplified before sequencing, and because it relies on many short overlapping reads, each section of DNA or RNA is sequenced multiple times. Also, because it is so much quicker and cheaper, it is possible to do more repeats than with Sanger sequencing. More repeats mean greater coverage, which leads to a more accurate and reliable sequence, even if individual reads are less accurate for NGS.
The nanopore and single molecule, real-time (SMRT) sequencing were recently employed to complete the sequence for the full human genome (2022). Previous genomic sequences were missing regions with highly repetitive sequences at the centromeres and telomeres. The "Telomere-to-Telomere (T2T) Consortium performed the analysis which added 200 megabases of new sequence information that was missing from the previous best sequence.
DNA Synthesis Techniques
DNA synthesis is the natural or artificial creation of deoxyribonucleic acid (DNA) molecules. The term DNA synthesis can refer to DNA replication (which will be covered in more detail in Chapter XX), polymerase chain reaction (PCR), or gene synthesis (physically creating artificial gene sequences).
Polymerase Chain Reaction (PCR)
The Polymerase chain reaction (PCR) refers to a technique employed widely in the basic and biomedical sciences. PCR is a laboratory technique utilized to amplify specific segments of DNA for a wide range of laboratory and/or clinical applications. Building on the work of Panet and Khorana’s successful amplification of DNA in-vitro, Kary Mullis and coworkers developed PCR in the early 1980s, having been met with a Nobel prize only a decade later. Allowing for more than the billion-fold amplification of specific target regions, it has become instrumental in many applications including the cloning of genes, the diagnosis of infectious diseases, and the screening of prenatal infants for deleterious genetic abnormalities.
Fundamentals
The main components of PCR are a template, primers, free nucleotide bases, and the DNA polymerase enzyme. The DNA template contains the specific region that you wish to amplify, such as the DNA extracted from a piece of hair for example. Primers, or oligonucleotides, are short strands of single-stranded DNA complementary to the 3' end of each target region. Both a forward and a reverse primer are required, one for each complementary strand of DNA. DNA polymerase is the enzyme that carries out DNA replication. Thermostable analogs of DNA polymerase I, such as Taq polymerase, which was originally found in a bacterium that grows in hot springs, are a common choice due to their resistance to the heating and cooling cycles necessary for PCR.
PCR takes advantage of the complementary base pairing, double-stranded nature, and melting temperature of DNA molecules. This process involves cycling through 3 sequential rounds of temperature-dependent reactions: DNA melting (denaturation), annealing, and enzyme-driven DNA replication (elongation). Denaturation begins by heating the reaction to about 95oC, disrupting the hydrogen bonds that hold the two strands of template DNA together. Next, the reaction is reduced to around 50 to 65oC, depending on the physicochemical variables of the primers, enabling the annealing of complementary base pairs. The primers, which are added to the solution in excess, bind to the beginning of the 3' end of each template strand and prevent re-hybridization of the template strand with itself. Lastly, enzyme-driven DNA replication, or elongation, begins by setting the reaction temperature to the amount which optimizes the activity of DNA polymerase, which is around 75 to 80oC. At this point, DNA polymerase, which needs double-stranded DNA to begin replication, synthesizes a new DNA strand by assembling free nucleotides in solution in the 3' to 5' direction to produce 2 full sets of complementary strands. The newly synthesized DNA is now identical to the template strand and will be used as such in the progressive PCR cycles. The steps in PCR are animated in the video below.
Figure (\PageIndex{17}\) below illustrate the step involved in PCR amplification of target DNA.

Figure (\PageIndex{18}\) shows a video animation of a PCR reaction.
Figure (\PageIndex{18}\): Video animation of a PCR reaction.
Given that previously synthesized DNA strands serve as templates, the amplification of DNA using PCR increases at an exponential rate, where the copies of DNA double at the end of each replication step. The exponential replication of the target DNA eventually plateaus around 30 to 40 cycles mainly due to reagent limitation, but can also be due to inhibitors of the polymerase reaction found in the sample, self-annealing of the accumulating product, and accumulation of pyrophosphate molecules.
Real-Time PCR
At its advent, PCR technology was limited to qualitative and or semi-quantitative analysis due to limitations on the ability to quantitate nucleic acids. At that time, to verify if the target gene was amplified successfully, the DNA product was separated by size via agarose gel electrophoresis. Ethidium bromide, a molecule that fluoresces when bound to dsDNA, could give a rough estimate of DNA amount by roughly comparing the brightness of separated bands, but was not sensitive enough for rigorous quantitative analysis.
Improvements in fluorophore development and instrumentation led to thermocyclers that no longer required measurement of only end-product DNA. This process, known as real-time PCR, or quantitative PCR (qPCR), has allowed for the detection of dsDNA during amplification. qPCR thermocyclers are equipped with the ability to excite fluorophores at specific wavelengths, detect their emission with a photodetector, and record the values. The sensitive collection of numerical values during amplification has strongly enhanced quantitative analytical power.
There are two main types of fluorophores used in qPCR: those that bind specifically to a given target sequence and those that do not. The sensitivity of fluorophores has been an important aspect of qPCR development. One of the most effective and widely used non-specific markers, SYBR Green, after binding to the minor groove of dsDNA, exhibits a 1000-fold increase in fluorescence compared to being free in solution (Video 5.1). However, if even more specificity is desired, a sequence-specific oligonucleotide, or hybridization probe, can be added, which binds to the target gene at some point in front of the primer (after the 3' end). These hybridization probes contain a reporter molecule at the 5' end and a quencher molecule at the 3' end. The quencher molecule effectively inhibits the reporter from fluorescing while the probe is intact. However, upon contact with DNA polymerase I, the hybridization probe is cleaved, allowing for the fluorescence of the dye (Video 5.1).
Reverse-Transcription PCR
Since its advent, PCR technology has been creatively expanded upon, and reverse-transcription PCR (RT-PCR) is one of the most important advances. Real-time PCR is frequently confused with reverse-transcription PCR, but they are separate techniques. In RT-PCR, the DNA amplified is derived from mRNA by using reverse-transcriptase enzymes, to produce a cDNA copy of the gene. Using primer sequences for genes of interest, traditional PCR methods can be used with the cDNA to study the expression of genes qualitatively. Currently, reverse-transcription PCR is commonly used with real-time PCR, which allows one to quantitatively measure the relative change in gene expression across different samples.
Figure (\PageIndex{19}\) shows a video animation video showing the use of reverse transcription Polymerase Chain Reaction (RT-PCR) in COVID-19 testing.
Figure (\PageIndex{19}\): Video animation of the Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Issues of Concern
One disadvantage of PCR technology is that it is extremely sensitive. Trace amounts of RNA or DNA contamination in the sample can produce extremely misleading results. Another disadvantage is that the primers designed for PCR require sequence data, and therefore can only be used to identify the presence or absence of a known pathogen or gene. Another limitation is that sometimes the primers used for PCR can anneal non-specifically to similar sequences, but not identical, to the target gene.
Another potential issue of using PCR is the possibility of primer dimer (PD) formation. PD is a potential by-product and consists of primer molecules that have hybridized with each other due to the strings of complementary bases in the primers. The DNA polymerase amplifies the PD, leading to competition for PCR reagents that could be used to amplify the target sequences.
Clinical Significance
PCR amplification is an indispensable tool with various applications within medicine. Often, it is used to test for the presence of specific alleles, such as in the case of prospective parents screening for genetic carriers, but it can also be used to diagnose the presence of disease directly and for mutations in the developing embryo. For example, the first time PCR was used in this way was for the diagnosis of sickle cell anemia through the detection of a single gene mutation.
Additionally, PCR has greatly revolutionized the diagnostic potential for infectious diseases, as it can be used to rapidly determine the identity of microbes that were traditionally unable to be cultured, or that required weeks for growth. Pathogens routinely detected using PCR include Mycobacterium tuberculosis, human immunodeficiency virus, herpes simplex virus, syphilis, and countless other pathogens. Moreover, qPCR is not only used for testing the qualitative presence of microbes but also to quantify bacterial, fungal, and viral loads.
The sensitivity of diagnostic tools for mutations to oncogenes and tumor suppression genes has been improved at least 10,000-fold due to PCR, allowing for earlier diagnosis of cancers like leukemia. PCR has also enabled more nuanced and individualized therapies for cancer patients. Additionally, PCR can be used for the tissue typing done that is vital to organ implantation and has even been proposed as a replacement for antibody-based tests for blood type. PCR also has clinical applications in the field of prenatal testing for various genetic diseases and/or clinical pathologies. Samples are obtained either via amniocentesis or chorionic villus sampling.
In forensic medicine, short pieces of repeating, highly polymorphic DNA, coined short tandem repeats (STRs), are amplified and used to compare specific variations within genes to differentiate individuals.[9] Primers specific for the loci of these STRs are used and amplified using PCR. Various loci contain STRs in the human genome, and the statistical power of this technique is enhanced by checking multiple sites.
Gene Synthesis
Artificial gene synthesis, sometimes known as DNA printing is a method in synthetic biology that is used to create artificial genes in the laboratory. Based on solid-phase DNA synthesis, it differs from molecular cloning and polymerase chain reaction (PCR) in that it does not have to begin with preexisting DNA sequences. Therefore, it is possible to make a completely synthetic double-stranded DNA molecule with no apparent limits on either nucleotide sequence or size.
The method has been used to generate functional bacterial or yeast chromosomes containing approximately one million base pairs. Creating novel nucleobase pairs in addition to the two base pairs in nature could greatly expand the genetic code.
Synthesis of the first complete gene, a yeast tRNA, was demonstrated by Har Gobind Khorana and coworkers in 1972. Synthesis of the first peptide- and protein-coding genes was performed in the laboratories of Herbert Boyer and Alexander Markham, respectively.
Commercial gene synthesis services are now available. Approaches are most often based on a combination of organic chemistry and molecular biology techniques and entire genes may be synthesized "de novo", without the need for template DNA. Gene synthesis is an important tool in many fields of recombinant DNA technology including heterologous gene expression, vaccine development, gene therapy, and molecular engineering. The synthesis of nucleic acid sequences can be more economical than classical cloning and mutagenesis procedures. It is also a powerful and flexible engineering tool for creating and designing new DNA sequences and protein functions.
Gene optimization
While the ability to make increasingly long stretches of DNA efficiently and at lower prices is a technological driver of this field, increasing attention is being focused on improving the design of genes for specific purposes. Early in the genome sequencing era, gene synthesis was used as an (expensive) source of cDNAs that were predicted by genomic or partial cDNA information but were difficult to clone. As higher-quality sources of sequence-verified cloned cDNA have become available, this practice has become less urgent.
Producing large amounts of protein from gene sequences can sometimes prove difficult. Many of the most interesting proteins are normally regulated to be expressed in very low amounts in wild-type cells. Redesigning these genes offers a means to improve gene expression in many cases. Rewriting the open reading frame is possible because of the degeneracy of the genetic code. Thus it is possible to change up to about a third of the nucleotides in an open reading frame and still produce the same protein. The available number of alternate designs possible for a given protein is astronomical. For a typical protein sequence of 300 amino acids, there are over 10150 codon combinations that will encode an identical protein. Codon optimization, or replacing rarely used codons with more common codons sometimes has dramatic effects. Further optimizations such as removing RNA secondary structures can also be included. At least in the case of E. coli, protein expression is maximized by predominantly using codons corresponding to tRNA that retain amino acid charging during starvation. Computer programs are used to optimize this task. A well-optimized gene can improve protein expression 2 to 10-fold, and in some cases, more than 100-fold improvements have been reported. Because of the large number of nucleotide changes made to the original DNA sequence, the only practical way to create the newly designed genes is to use gene synthesis.
Oligonucleotide synthesis
Oligonucleotides are chemically synthesized using building blocks called nucleoside phosphoramidites. These can be normal or modified nucleosides that have protecting groups to prevent their amines, hydroxyl groups, and phosphate groups from interacting incorrectly. One phosphoramidite is added at a time, the 5' hydroxyl group is deprotected and a new base is added, and the process is repeated. The chain grows in the 3' to 5' direction, which is backward relative to DNA biosynthesis in vivo. In the end, all the protecting groups are removed. The solid phase DNA synthesis reaction is shown in Figure (\PageIndex{20}\) below.

Nevertheless, being a chemical process, several incorrect interactions occur leading to some defective products. The longer the oligonucleotide sequence that is being synthesized, the more defects there are, thus this process is only practical for producing short sequences of nucleotides. The current practical limit is about 200 bp (base pairs) for an oligonucleotide with sufficient quality to be used directly for a biological application. HPLC can be used to isolate products with the proper sequence. Meanwhile, a large number of oligos can be synthesized in parallel on gene chips. For optimal performance in subsequent gene synthesis procedures, they should be prepared individually and on larger scales.
DNA synthesis and synthetic biology
The significant drop in the cost of gene synthesis in recent years due to increasing competition of companies providing this service has led to the ability to produce entire bacterial plasmids that have never existed in nature. The field of synthetic biology utilizes the technology to produce synthetic biological circuits, which are stretches of DNA manipulated to change gene expression within cells and cause the cell to produce a desired product.
The ability to synthetically produce DNA will enable the development of environmental, medical, and commercially relevant products. For example, in 2015, Novartis, in collaboration with the Synthetic Genomics Vaccines inc. and the US Biomedical Advanced Research and Development Authority, announced that they had effectively created a synthetic DNA influenza vaccine. New synthetic DNA vaccines hold promise to provide an alternative to current egg-produced conventional vaccines that can be plagued by low efficacy.
DNA vaccines can avoid many issues associated with egg-based vaccine production by generating viral proteins within host cells. To create a DNA vaccine, an antigen-encoding gene is cloned into a non-replicative expression plasmid, which is delivered to the host by traditional vaccination routes. Host cells that take up the plasmid express the vaccine antigen which can be presented to immune cells via the major histocompatibility complex (MHC) pathways. CD4+ T helper cell activation following MHC class II presentation of secreted DNA vaccine protein is critical for the production of antigen-specific antibodies as shown in Figure (\PageIndex{21}\) below.

After two decades of research, DNA vaccine technology is gaining maturity—several veterinary DNA vaccines are currently licensed for West Nile virus and melanoma, and significantly, the first commercial DNA vaccine against H5N1 in chickens has recently been conditionally approved by the USDA. In addition, ongoing large animal trials of DNA vaccines against other diseases such as HIV, hepatitis, and Zika virus offer valuable insights that can be applied to influenza DNA vaccine design. Promising approaches have arisen from the numerous studies evaluating different DNA vaccine formulations and delivery systems, but a strategy that consistently elicits protection against influenza in large animal models has not yet emerged. Successful plasmid delivery and the use of appropriate adjuvants remain key challenges that need to be addressed before influenza DNA vaccines become effective for human use.
RNA Vaccines
Of course, amazing progress has been made in the creation of RNA vaccines as demonstrated by the Moderna and Pfizer RNA vaccines to the spike protein of the SARS-CoV2 virus. Models show that just in the US through 2022, Covid vaccines saved the US $1.15 trillion and 3 million lives. Just in the first year, the vaccines are estimated to have saved upwards of 20 million lives worldwide, an accomplishment worthy of every prize in the world! Many more could have been saved in the developing world if the vaccine was more widely available.
As we learned previously, RNA is much more labile to hydrolysis than DNA since RNA has a 2' OH group. Methods to stabilize RNA were required before an RNA vaccine could become a reality. More importantly, dsRNA is a danger signal that a viral infection may be present. dsRNA, a pathogen-associated molecular pattern (PAMP), binds to the Toll-like Receptor 3 (TLR3) and initiates an inflammatory response that would eliminate the RNA before it could be decoded to form a protein sequence that could elicit an antibody response, a requirement for a vaccine.
Katalin Karikó and Drew Weissman found that modifying the uracil bases to pseudouracil (Ψ) prevents the PAMP response and allows RNA to last long enough to create the protein sequence (i.e. it increases the stability of the RNA to hydrolysis). Pseudouracil (Ψ) is found in structural RNAs (transfer, ribosomal, small nuclear, and small nucleolar), and in fact is the most common modification found in RNA. It even is metabolized by a naturally occurring pathway back to uracil.
Figure (\PageIndex{22}\) below shows the structures of uracil and the N1-methyl derivative of pseudouracil attached to ribose in an RNA and their base pairing to adenine. The N1-methylpseudouracil base still bases pairs with an adenine base. Hence the RNA structure and its functional ability to be decoded into a protein sequence is not affected by the modification. In short, mRNAs modified to contain pseudouracil have a much greater translational capacity as well as stability. In addition, methylation of uracil decreases the immunogenicity of RNAs that contain it.

Figure (\PageIndex{23}\) below shows a cartoon version of the modified mRNA (a) Covid-19 and its encapsulation into a lipid nanoparticle (b) for an mRNA vaccine.

Panel (a) shows the generalized structure of the mRNA vaccine for the S gene which encodes the surface spike protein of the virus. Panel b shows the lipid nanoparticle that encapsulates and protects the mRNA vaccine. The lipids include proprietary mixtures of phospholipids, cholesterol, cationic (ionizable) lipids, and polyethylene glycol (PEGylated). Without the basic research of Karikó and Weissman (and many others), the world would not have had the Covid-19 vaccine in time to save so many lives. They were awarded the Nobel Prize in Medicine in 2023 for their work.
The biosynthesis of pseudouridine, the most common modification of cellular RNA, is done after DNA transcription (i.e. post-transcriptionally) using the enzyme pseudouridine synthases (PUS), which is found in all kingdoms in life. The reaction involves
- cleavage of the C-N-glycosidic bond of uridine in RNA
- rotation of the cleaved uracil to align C5 of uracil and C1′ of the ribose
- formation of the C1′-C5 carbon–carbon bond.
These processes are illustrated in Figure (\PageIndex{24}\) below.
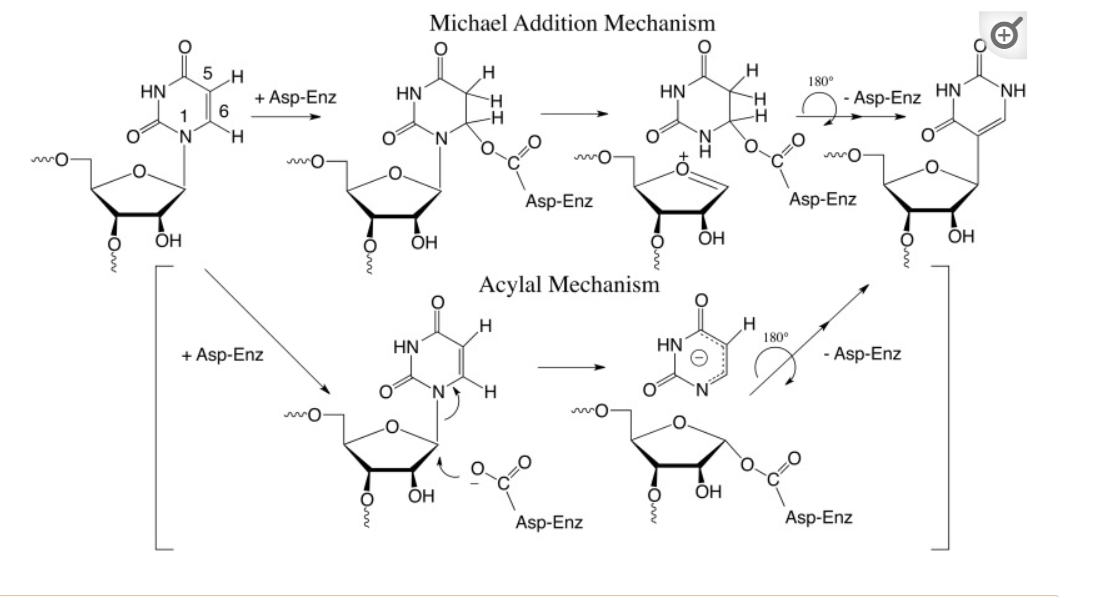
Figure (\PageIndex{24}\): Post-transcriptional modification of uridine to pseudouridine. Czudnochowski N. et al. The mechanism of pseudouridine synthases from a covalent complex with RNA, and alternate specificity for U2605 versus U2604 between close homologs. Nucleic Acids Res. 2014 Feb;42(3):2037-48. doi: 10.1093/nar/gkt1050. Epub 2013 Nov 7. PMID: 24214967; PMCID: PMC3919597. Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0/)