
9.8: Electrophoresis
- Page ID
- 3085

DNA molecules are long and loaded with negative charges, thanks to their phosphate backbones. Electrophoretic methods separate large molecules, such as DNA, RNA, and proteins based on their charge and size. For DNA and RNA, the charge of the nucleic acid is proportional to its size (length). For proteins, which do not have a uniform charge, a clever trick is employed to make them mimic nucleic acids.

Agarose
Agarose gel electrophoresis is a method for separating nucleic acids. It is worth noting that nucleic acids are the largest molecules found in cells, in some cases by orders of magnitude. Agarose provides a matrix which encases a buffer. The matrix provides openings for macromolecules to move through and the largest macromolecules have the most difficult time navigating, whereas the smallest macromolecules slip through it the easiest. Unlike column chromatography, electrophoresis uses an electric current as a force to drive the molecules through the matrix. Since the size to charge ratios for DNA and RNA are constant for all sizes of these nucleic acids, the size per force is also constant (since force is directly proportional to charge), so the molecules simply sort on the basis of their size - the smallest move fastest and the largest move slowest. Visualization of the DNA fragments in the gel is made possible by addition of a dye, such as ethidium bromide that fluoresces under ultraviolet light.

SDS-PAGE
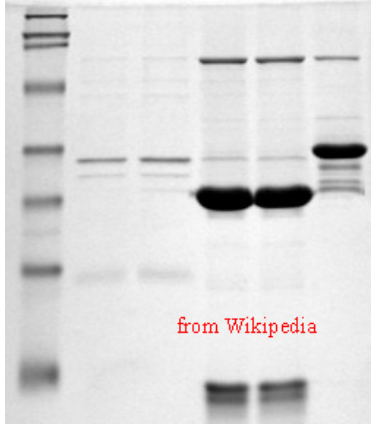
Like DNA and RNA, proteins are large macromolecules. Proteins, however, vary tremendously in their charge. Whereas double-stranded DNA is rod-shaped, most proteins are globular (folded up). Further, proteins are considerably smaller than nucleic acids, so the openings of the matrix of the agarose gel are simply too large to effectively provide separation. Consequently, unlike nucleic acids, proteins cannot be effectively separated by electrophoresis on agarose gels. To separate proteins by electrophoresis, one must make several modifications. First, a matrix made by polymerizing and crosslinking acrylamide units is employed. One can adjust the pore size of the matrix readily by changing the percentage of acrylamide in the gel. Higher percentages of acrylamide create smaller pores and are more effective in separating smaller molecules, whereas lower percentages of acrylamide reverse that. Second, proteins must be physically altered to “present" themselves to the matrix like the negatively charged rods of DNA. This is accomplished by treating the proteins with the detergent called SDS (sodium dodecyl sulfate). SDS denatures the proteins so they assume a rod-like shape and the SDS molecules coat the proteins such that the exterior surface is loaded with negative charges proportional to the mass, just like the backbone of DNA. Third, a “stacking gel" may be employed at the top of the gel to provide a way of compressing the samples into a tight band before they enter the main polyacrylamide gel (called the resolving gel). Just as DNA fragments get sorted on the basis of size (largest move slowest and smallest move fastest), the proteins migrate through the gel matrix at rates inversely related to their size. Upon completion of the electrophoresis, there are several means of staining to visualize the proteins on the gel. They include reagents, such as Coomassie Brilliant Blue or silver nitrate (the latter is much more sensitive than Coomassie Blue staining and can be used when there are very small quantities of protein).
Isoelectric Focusing
Proteins vary considerably in their charges and, consequently, in their pI values (pH at which their charge is zero). Separating proteins by isoelectric focusing requires establishment of a pH gradient in an acrylamide gel matrix. The matrix’s pores are adjusted to be large to reduce the effect of sieving based on size. Molecules to be focused are applied to the gel with the pH gradient and an electric current is passed through it. Positively charged molecules, for example, move towards the negative electrode, but since they are traveling through a pH gradient, as they pass through it, they reach a region where their charge is zero and, at that point, they stop moving. They are at that point attracted to neither the positive nor the negative electrode and are thus “focused" at their pI. By using isoelectric focusing, it is possible to separate proteins whose pI values differ by as little as 0.01 units.
2-D Gel Electrophoresis
Both SDS-PAGE and isoelectric focusing are powerful techniques, but a clever combination of the two is a powerful tool of proteomics - the science of studying all of the proteins of a cell/tissue simultaneously. In 2D gel electrophoresis, an extract containing the proteins is first prepared. One might, for example, be studying the proteins of liver tissue. The liver cells are lysed and all of the proteins are collected into a sample. Next, the sample is subjected to isoelectric focusing as described earlier, to separate the proteins by their pI values. Next, as shown on the previous page, the isoelectric gel containing the separated proteins is rotated through 90º and placed on top of a regular polyacrylamide gel for SDS-PAGE analysis (to separate them based on size). The proteins in the isoelectric gel matrix are electrophoresed into the polyacrylamide gel and separation on the basis of size is performed. The product of this analysis is a 2D gel, in which proteins are sorted by both mass and charge.

The power of 2D gel electrophoresis is that virtually every protein in a cell can be separated and appear on the gel as a distinct spot. In the figure, spots in the upper left correspond to large positively charged proteins, whereas those in the lower right are small negatively charged ones. It is possible using high- throughput mass spectrometry analysis to identify every spot on a 2D gel. This is particularly powerful when one compares protein profiles between different tissues or between the samples of the same tissue treated or untreated with a particular drug. Comparison of a 2D separation of a non-cancerous tissue with a cancerous tissue of the same type provides a quick identification of proteins whose level of expression differs between them. Information such as this might be useful in designing treatments or in determining the mechanisms by which the cancer arose.