
3.3: Proteins
- Page ID
- 2970

Whereas nucleotides all are water soluble and have the same basic composition (sugar, base, phosphate) and the sugars also are water soluble and mostly contain 5 or 6 carbons (a few exceptions), the amino acids (general structure below) are structurally and chemically diverse.

Though all of the amino acids are, in fact, soluble in water, the interactions of their side chains with water differ significantly. This is important, because it is only in the side chains (R-groups) that amino acids differ from each other. Based on side chains, we can group the 20 amino acids found in proteins as follows:
- Aromatic (phenylalanine, tyrosine, tryptophan)
- Aliphatic (leucine, isoleucine, alanine, methionine, valine)
- Hydroxyl/Sulfhydryl (threonine, serine, tyrosine, cysteine)
- Carboxyamide (glutamine, asparagine)
- R-Acids (glutamic acid, aspartic acid)
- R-Amines (lysine, histidine, arginine)
- Odd (glycine, proline)
Note that tyrosine has a hydroxyl group and fits into two categories. Note also that biochemistry books vary in how they organize amino acids into categories. Amino acids are joined to each other by peptide bonds. This introduces a slight simplifying aspect to the structure of proteins – one need only consider the positioning of the R-groups around each peptide bond when determining protein structure schematically. Proteins that are in aqueous environments, such as the cytoplasm of the cell, have their amino acids arranged so that those with hydrophilic side chains (such as threonine or lysine) predominate on the exterior of the protein so as to interact with water. The hydrophobic amino acids in these proteins are found predominantly on the interior. When one examines the structure of proteins in non-aqueous environments, such as the interior of a lipid bilayer, the arrangement is flipped – hydrophobics predominate on the outside where they can interact with the hydrophobic side chains of membrane fatty acids and the hydrophilic amino acids are arranged anyplace where they can contact water. For a protein like porin, which provides an interior channel through which water can pass, this is where the hydrophilics are found. For transmembrane proteins, which project through both sides of the membrane, the hydrophilics are found at each point where the polypeptide chain emerges from the membrane.

Primary Structure
How do proteins obtain such arrangements of amino acids? As we shall see, the structures of all proteins ultimately arise from their amino acid sequences. The amino acid sequence is referred to as the primary structure and changes in it can affect every other level of structure as well as the properties of a protein. The primary structure of a protein arrived at its current state as a result of mutation and selection over evolutionary time. On a more immediate time scale, 3D protein structure arises as a result of a phenomenon called folding. Protein folding results from three different structural elements beyond primary structure. They are referred to as secondary, tertiary, and quaternary structures, each arising from interactions between progressively more distant amino acids in the primary structure.

Secondary Structure
Interactions between amino acids within about ten units of each other give rise to regular repeating structures. These secondary structures include the well known alpha-helix and beta strands. Both were predicted by Linus Pauling, Robert Corey, and Herman Branson in 1951. Each structure has unique features. We use the terms rise, repeat, and pitch to describe the parameters of a helix. The repeat is the number of residues in a helix before it begins to repeat itself. The rise is the distance the helix elevates with addition of each residue. The pitch is the distance between the turns of the helix.
Alpha Helix
The alpha helix (Figure 3.1.3) forms as the result of interactions between amino acids separated by four residues. Interesting;y, the side chains of the amino acids in an alpha helix are all pointed outwards from the axis of the helix. Alpha helices have a repeat of 3.6 amino acid residues per turn of the helix, meaning that four turns of the helix have approximately 14 amino acid residues. Hydrogen bonds occur between the C=O of one amino acid and the N-H of another amino acid four residues distant and these help to stabilize the structure (note that the C=O and N-H involved are part of the polypeptide backbone, not the R-groups). Some amino acids have high helix forming tendencies. They include methionine, alanine, leucine, uncharged glutamate, and lysine. Others, such as proline, glycine, and negatively charged aspartate, disfavor its formation.
Beta Strands
Beta strands are the most fundamental helix, having essentially a 2D backbone of 'fold' like those of the pleats of a curtain. Indeed, beta strands can be arranged together to form what are called beta sheets. Other regular structures are also known. What determines whether a given stretch of a protein is in a helical or other structure? Here is where the shape and chemistry of the side chains play a role.

Fibrous Proteins
Not all proteins have significant amounts of tertiary or quaternary structure. (As we shall see, these last two levels of structure arise from 'bends' in polypeptide chains and interactions between separate polypeptide chains, respectively.)
Alpha keratin, for example, is what we refer to as a fibrous protein (also called scleroprotein). Alpha keratin has primary structure and secondary structure, but little tertiary or quaternary structure.
Consequently, alpha keratin exists mostly as long fibers, such as are found in hair. Beta-keratin is a harder fibrous protein found in nails, scales, and claws. It is made up mostly of beta sheets. Proline, which is the least flexible amino acid, due to attachment of the side chain to the alpha-amino grip, is less likely to be found in alpha helices, but curiously it is found abundantly in the fibrous protein known as collagen. Collagen (previous page) is the most abundant protein in the human body and is the 'glue' that literally sticks us together. How does the inflexibility of proline permit it to be in a helix? The answer is probably the parallel abundance in collagen of glycine, which contains the smallest side group and therefore has the greatest flexibility.
As an interesting sidelight of the presence of proline in collagen is the chemical modification of prolines, by the addition of hydroxyl groups, after the protein is made. Such 'post-translational modifications' are not uncommon. Threonine, serine, and tyrosine frequently have their hydroxyl side-chains phosphorylated. Lysines in collagen too are hydroxylated post-translationally. The hydroxylated prolines and lysines play a role in the formation of interchain hydrogen bonds and crosslinking of triple helices during the assembly of collagen fibrils. These bonds provide structural integrity to the collagen. The enzymes that add hydroxyls to proline and lysine require vitamin C (ascorbic acid) for their activity. Lack of vitamin C leads to the production of weakened collagen fibrils, resulting in a condition called scurvy.
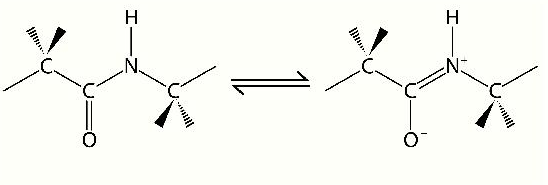
The carbonyl oxygen of the peptide bond can exist in resonance with the C-N bond, giving the peptide bond characteristics of a double bond and imposing limitations for rotation around it. If we trat the peptide bond as a double bond, then the arrangements of adjacent carbon bonds around it can be thought of as being in the cis or trans configurations. In proteins, not surprisingly, the preferred arrangement of these groups is strongly trans (1000/1). Of the 20 amino acids, the one that favors peptide bonds in the cis configuration most commonly is proline, but even for proline, the trans isomer is strongly preferred.
Figure 3.2.5: Collagen.
Ramachandran Plots
Another consequence of considering the peptide bond as a double bond is that it reduces the number of variable rotational angles of the polypeptide backbone. The terms phi and psi refer to rotational angles about the bonds between the N-alpha carbon and alpha carbon-carbonyl carbon respectively (previous page). Given the bulkiness of R-groups, the phenomenon of steric hindrance and the tendency of close side chains to interact with each other, one might expect to find a bias in the values of phi and psi. Indeed, that is exactly what is observed. Dr. G.N. Ramachandran proposed such a result and, in a plot that bears his name, depicted the theoretical likelihood of each angle appearing in a polypeptide. More recent observations of actual phi and psi angles in data from the PDB protein database bear out Dr. Ramachandran's predictions. In the plot above, beta strands fit nicely in the darker blue section at the top and alpha helices fit in the yellow section near the middle.


Tertiary Structure
In contrast to secondary structures, which arise from interactions between amino acids close in primary structure, tertiary structure arises from interactions between amino acids more distant in primary structure. Such interactions are not possible in an endlessly stretching fiber because each amino acid placed between two amino acids causes them to be moved farther away from each other in what is essentially the two dimensions of a secondary structure. For distant amino acids to interact, they must be brought into closer proximity and this requires bending and folding of the polypeptide chain. Proteins with such structures are referred to as ‘globular’ and they are, by far, the most abundant class of proteins. Indeed, it is in globular proteins that we have the most vivid images of the results of folding. “Folds" in polypeptides arise as a result of ‘bends’ between regions of secondary structure (such as alpha helix or beta strands). Such structures may be preferred due to incompatibility of a given amino acid side chain for a secondary structure formed by the amino acids preceding it. Bends occur commonly in proteins and proline is often implicated. Bends do not have the predictable geometry of alpha helices or beta strands and are often referred to as random coils. Thus, even though protein structure can be described easily as regions of secondary structure separated by bends, the variability of bend structures makes prediction of tertiary structure from amino acid sequence enormously more difficult than identifying/predicting regions of secondary structure.

Hydrophobic Effect
It is at the level of tertiary structure that the characteristic arrangement of hydrophobic and hydrophilic amino acids in a protein occurs. In an aqueous environment, for a protein to remain soluble, it must have favorable interactions with the water around it, hence, the positioning of hydrophilic amino acids externally. Another impetus for the folding phenomenon is a bit harder to understand. It is known as the hydrophobic effect. At a chemical level, it makes sense – hydrophobic amino acids will ‘prefer’ to interact with each other internally and away from water. The driving force for this phenomenon, though, is a bit more conceptually difficult. Consider a bottle containing oil and water. As everyone knows, the two liquids will not mix and instead will form separate layers. A reasonable question might be why they do this instead of one existing as tiny globules inside of the other. The answer to that question, as well as the positioning of hydrophobic amino acids in the interior of water soluble proteins, is the hydrophobic effect. To understand the hydrophobic effect, perform the followingexperiment – take the water-oil mixture and shake it vigorously. This will force the layers to mix and one will observe that tiny globules of both water and oil can, in fact, be found initially in the layer of each. Over time, though, the tiny globules break up and merge with the appropriate layer. This is due to the phenomenon of entropy and consideration of surface area. First, the sum of the surface area of the embedded tiny globs is far greater than the area of the region between the two layers after mixing is over. The smaller the globs, the more the surface area of interaction between the oil and the water. The minimum possible surface area of interaction occurs when there are no globs at all – just two layers and nothing else.

How does this relate to entropy? Interactions between the water-hydrophobic layers causes the molecules at the interface to arrange themselves precisely/regularly so as to minimize their interactions. Ordering thus occurs at the layer interfaces. The
maximum amount of ordering occurs when the maximum surface areas of oil and water interact. Small globules give rise to more
exposed surface area between the water and hydrophobic layers and, as a consequence, more ordering. Since entropy in a closed system tends to increase, it will tend to reduce the amount of ordering, if left alone. Thus, one can increase the ordering on a nanoscopic scale (forming globules) by applying energy in the form of shaking. When left alone, however, the system will increase its disorder by reducing the interactions between hydrophobic groups and hydrophilic ones.


In the oil water mixture, this causes the tiny globs to break up and produce the two layers we are familiar with because this is the
minimum surface area that can be made between the two layers and thus the least ordering. In proteins, hydrophobic amino acid side chains are ‘shielded’ from water by placement internal to the protein, thus also reducing interfaces between hydrophobic residues and water. In both cases, entropy is increased, due to the reduced organization of the layers. Once formed, the interactions between the hydrophobic amino acid side chains helps to stabilize the overall protein structure.


Quaternary Structure
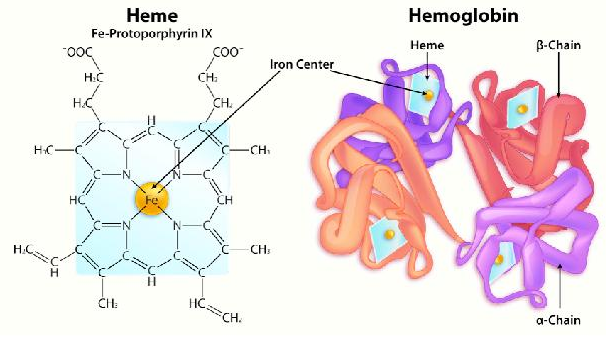
The last level of protein structure we will consider is that of quaternary structure. In order to have quaternary structure, a protein must have multiple polypeptide subunits because the structure involves the arrangement of those subunits with respect to each other. Consider hemoglobin, the oxygen-carrying protein of our blood. It contains two identical subunits known as alpha and two other identical ones known as beta. These are arranged together in a fashion as shown on the previous page. By contrast, the related oxygen storage protein known as myoglobin only contains a single subunit. Hemoglobin has quaternary structure, but myoglobin does not. Multiple subunit proteins are common in cells and they give rise to very useful properties not found in single subunit proteins. In the case of hemoglobin, the multiple subunits confer the property of cooperativity – variable affinity for oxygen depending on the latter’s concentration. In the case of enzymes, it can impart allosterism – the ability to have the activity of the enzyme altered by interaction with an effector molecule. We will discuss allosterism in detail in the next chapter.
Other Protein Structural Features
Not everything found in a protein is an amino acid. Proteins frequently have other chemical groups, known as prosthetic groups, bound to them, that are necessary for the function of a protein. Examples include the porphyrin ring of heme in myoglobin and hemoglobin that carries an iron so that oxygen can be bound. Metals are frequently employed by enzymes in their catalysis. Several vitamins (referred to as coenzymes), such as thiamine (B1) and riboflavin(B2) are modified and chemically bound to enzymes to help them perform specific catalytic functions.
Cooperativity
An interesting and important aspect of some proteins is the phenomenon of cooperativity. Cooperativity refers to the fact that binding of one ligand molecule by a protein favors the binding of additional molecules of the same type. Hemoglobin, for example, exhibits cooperativity when the binding of an oxygen molecule by the iron of the heme group in one of the four subunits causes a slight conformation change in the subunit. This happens because the heme iron is attached to a histidine side chain and binding of oxygen ‘lifts’ the iron along with the histidine ring (also known as the imidazole ring).
Since each hemoglobin subunit interacts with and influences the other subunits, they too are induced to change shape slightly when the first subunit binds to oxygen (a transition described as going from the T-state to the R-state). These shape changes favor each of the remaining subunits binding oxygen, as well. This is very important in the lungs where oxygen is picked up by hemoglobin, because the binding of the first oxygen molecule facilitates the rapid uptake of more oxygen molecules. In the tissues, where the oxygen concentration is lower, the oxygen leaves hemoglobin and the proteins .ips from the R-state back to the T-state.
Cooperativity is only one of many fascinating structural aspects of hemoglobin that help the body to receive oxygen where it is
needed and pick it up where it is abundant. Hemoglobin also assists in the transport of the product of cellular respiration (carbon dioxide) from the tissues producing it to the lungs where it is exhaled. Let us consider these individually.
Bohr Effect
The Bohr Effect was first described over 100 years ago by Christian Bohr. Shown graphically (above left), the observed effect is that hemoglobin’s affinity for oxygen decreases as the pH decreases and/or as the concentration of carbon dioxide increases. Binding of the protons by histidine helps to facilitate structural changes in the protein and also with the uptake of carbon dioxide. Physiologically, this has great significance because actively respiring tissues (such as contracting muscles) require oxygen and release protons and carbon dioxide. The higher the concentration of protons and carbon dioxide, the more oxygen is released to feed the tissues that need it most.

2,3 BPG
Another molecule affecting the release of oxygen by hemoglobin is 2,3 bisphosphoglycerate (also called 2,3 BPG or just BPG). Like protons and carbon dioxide, 2,3 BPG is produced by actively respiring tissues, as a byproduct of glucose metabolism. The 2,3 BPG molecule fits into the ‘hole of the donut’ of adult hemoglobin. Such binding of 2,3 BPG favors the T (tight) state of hemoglobin, which has a reduced affinity for oxygen. In the absence of 2,3 BPG, hemoglobin can exist in the R (relaxed) state, which has a high affinity for oxygen.
Fetal Hemoglobin
Adult hemoglobin releases oxygen when it binds 2,3 BPG. This is in contrast to fetal hemoglobin, which has a slightly different configuration (α2Γ2) than adult hemoglobin (α2ß). Fetal hemoglobin has a greater affinity for oxygen than maternal hemoglobin, allowing the fetus to obtain oxygen effectively from the mother’s blood. Part of the reason for fetal hemoglobin’s greater affinity for oxygen is that it doesn’t bind 2,3 BPG.
Another significant fact about 2,3 BPG is that its concentration is higher in the blood of smokers than it is of non-smokers. Consequently, hemoglobin in a smoker’s blood spends more time in the T state than the R state. That is a problem when it is in the lungs, where being in the R state is necessary to maximally load the hemoglobin with oxygen. A high blood level of 2,3 BPG is one of the reasons smokers have trouble breathing when they exercise – they have reduced oxygen carrying capacity.
Last, though it is not related directly to 2,3 BPG, smokers have another reason why their oxygen carrying capacity is lower than that of non-smokers. Cigarette smoke contains carbon monoxide and this molecule, which has almost identical dimensions to molecular oxygen, competes effectively with oxygen for binding to the iron atom of heme. Part of carbon monoxide’s toxicity is due to its ability to bind hemoglobin and prevent oxygen from binding.

Denaturation
For proteins, function is dependent on precise structure. Loss of the precise, folded structure of a protein is known as denaturation and is usually accompanied by loss of function. Anyone who has ever worked to purify an enzyme knows how easy it is for one to lose its activity. A few enzymes, such as ribonuclease, are remarkably stable under even very harsh conditions. For most others, a small temperature or pH change can drastically affect activity. The reasons for these differences vary, but relate to 1) the strength of the forces holding the structure together and 2) the ability of a protein to refold itself after being denatured. Let us consider these separately below.

Forces Stabilizing Structures
Amino acids are linked one to the other by peptide bonds. These covalent bonds are extraordinarily stable at neutral pHs, but can be broken by hydrolysis with heat under acidic conditions. Peptide bonds, however, only stabilize primary structure and, in fact, are the only relevant force responsible for it. Secondary structure, on the other hand, is generally stabilized by weaker forces, including hydrogen bonds. Hydrogen bonds are readily disrupted by heat, urea, or guanidinium chloride.
Forces stabilizing tertiary structure include ionic interactions, disulfide bonds, hydrophobic interactions, metallic bonds, and hydrogen
bonds. Of these, the ionic interactions are most sensitive to pH changes. Hydrophobic bonds are most sensitive to detergents. Thus,
washing one’s hands helps to kill bacteria by denaturing critical proteins they need to survive. Metallic bonds are sensitive to oxidation/reduction. Breaking disulfide bonds requires either a strong oxidizing agent, such as performic acid or a strong reducing agent
on another disulfide, such as mercaptoethanol or dithiothreitol.
Quaternary structures are stabilized by the same forces as tertiary structure and have the same sensitivities.
Refolding Denatured Proteins
All of the information for protein folding is contained in the primary structure of the protein. It may seem curious then that most proteins do not refold into their proper, fully active form after they have been denatured and the denaturant is removed. A few do, in fact, refold correctly under these circumstances. A good example is bovine ribonuclease (also called RNase). Its catalytic activity is very resistant to heat and urea. However, if one treats the enzyme with mercaptoethanol (which breaks disulfide bonds) prior to urea treatment and
heating, activity is lost, indicating that the covalent disulfide bonds help stabilize the overall enzyme structure. If one allows the enzyme
mixture to cool back down to room temperature, over time some enzyme activity reappears, indicating that ribonuclease can re-fold under the proper conditions.
Irreversible Denaturation
Most enzymes, however, do not behave like ribonuclease. Once denatured, their activity cannot be recovered to any significant extent. This may seem to contradict the idea that folding information is inherent to the sequence of amino acids in the protein. It does not. The reason most enzymes can’t refold properly is due to two phenomena. First, normal folding may occur as proteins are being made. Interactions among amino acids early in the synthesis are not “confused" by interactions with amino acids later in the synthesis because those amino acids aren’t present as protein synthesis starts. In many cases, the proper folding of newly made polypeptides is also assisted by special proteins called chaperones. Chaperones bind to newly made proteins, preventing interactions that might result in misfolding. Thus, early folding and the assistance of chaperones eliminate some potential “wrong-folding" interactions that can occur if the entire sequence was present when folding started.
Denatured full-length polypeptides have many more potential wrong folds that can occur. A second reason most proteins don’t refold properly after denaturation is probably that folding, like any other natural phenomenon, is driven by energy minimization. Though the folded structure may have a low energy, the path leading to it may not be all downhill. Like a chemical reaction that has energies of activation that must be overcome for the reaction to occur, folding likely has peaks and valleys of energy that do not automatically lead directly to the proper fold. Again, folding during synthesis leads the protein along a better-defined path through the energy maze of folding that denatured full-length proteins can’t navigate.
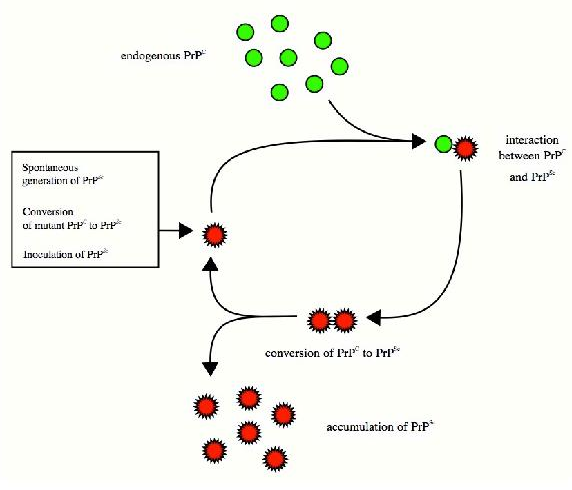
Prions and Misfolding
Folding and the stability of folded proteins is an important consideration for so-called “infectious" proteins known as prions. These mysterious proteins, which are implicated in diseases, such as mad cow disease and the related human condition known as Creutzfeldt-Jakob disease, result from the improper folding of a brain protein known as PrP. The misfolded protein has two important properties that lead to the disease. First, it tends to aggregrate into large complexes called amyloid plaques that damage/destroy nerve cells in the brain, leading ultimately to dementia and loss of brain function.
Second, and probably worse, the misfolded protein “induces" other copies of the same protein to misfold as well. Thus, a misfolded
protein acts something like a catalytic center and the disease progresses rapidly. The question arises as to how the PrP protein misfolds to begin with, but the answer to this is not clear. There are suggestions that exposure in the diet to misfolded proteins may
be a factor, but this is disputed. An outbreak of mad cow disease in Britain in the 1980s was followed by a rise in the incidence of a rare form of human Creutzfeld-Jakob disease called variant CJD (v-CJD), lending some credence to the hypothesis. It is possible that misfolding of many proteins occurs sporadically without consequence or observation, but if PrP misfolds, the results are readily apparent. Thus, Creutzfeld-Jakob disease may ultimately give insights into the folding process itself.
Dr. Kevin Ahern and Dr. Indira Rajagopal (Oregon State University)